Why do we get seemingly few SNPs with GBS data?
Methods: Used bash to count the occurrences of tags. Check your data and let me know what you find:
cat Demutliplexed.fastq | sed -n '2~4p' | cut -c4-70 | sort | uniq -c | grep -v N | sed 's/\s\s*/ /g' > TagCounts
Terminology: I have tried to use tags to refer to a particular sequence and reads as occurrences of those sequences. So a tag may be found 20 times in a sample (20 reads of that sequence).
Findings: Tag repeat distribution
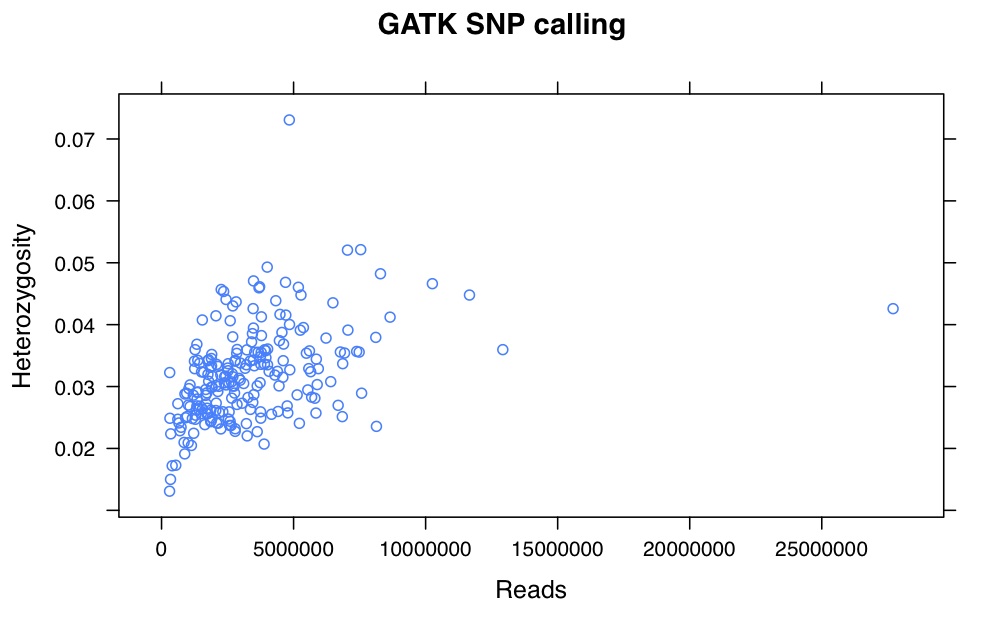
Probably the key thing and a big issue with GBS is the number of times each tag gets sequence. Most sites get sequenced once but most tags get sequence many many times. It is hard to see but here are histograms of the number of times each tags occur for a random sample (my pet11): All tags histogram (note how long the x axis is), 50 tags or less Histogram. Most tags occur once – that is the spike at 1. The tail is long and important. Although only one tag occurs 1088 times it ‘takes up’ 1088 reads.
How does this add up?
In this sample there are 3,453,575 reads. These reads correspond to 376,036 different tag sequences. This would mean (ideally) ~10x depth of each of the tags. This is not the case. There are a mere 718 tags which occur 1000 or more times but they account for 1394905 reads. That is 40% of the reads going to just over 700 (potential) sites. I’ve not looked summarized more samples using the same method but I am sure it would to yield the same result.
Here is an example: Random Deep Tag. Looking at this you can see that the problem is worse than just re-sequencing the same tag many times but you introduce a large number of tags that are sequencing and/or PCR errors that occur once or twice (I cut off many more occurances here).
Conclusion: Poor/incomplete digestion -> few sites get adapters -> they get amplified like crazy and then sequenced.
Update 1:
Of the > 3.4Million tags that are in the set of 18 samples I am playing with only 8123 are in 10 or more of the samples.
For those sites with >10 scored the number of times a tag is sequenced is correlated between samples. The same tags are being sequenced repeatedly in the different samples.
Update 2:
As requested the ‘connectivity’ between tags. This is the number of 1bp miss matches each tag has. To be included in this analysis a tag must occur at least 5 times in 3 individuals. Here is the figure. So most tags have zero matches a smaller number have one and so on. This actually looks pretty good – at least how I am thinking about it now. This could mean that the filtering criteria works. If errors were still being included I would expect tags with one match (the actual sequence) to occur more than ones with zero.