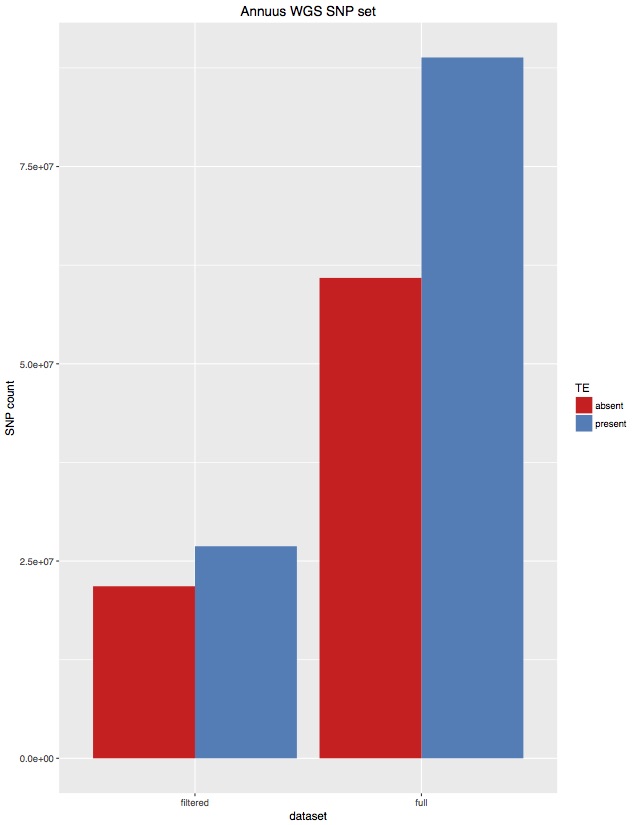
Considering the amount of sequencing coming out of the newer Illumina machines, we’ve started to combine GBS libraries with other samples. Due to how GBS libraries are made, when multiplexed with whole genome sequencing samples, there is an appreciable amount of contamination from GBS to WGS. That means you will find GBS reads in your WGS data. I’ve quantified that in the following figure, showing the percent of barcoded reads in WGS samples.
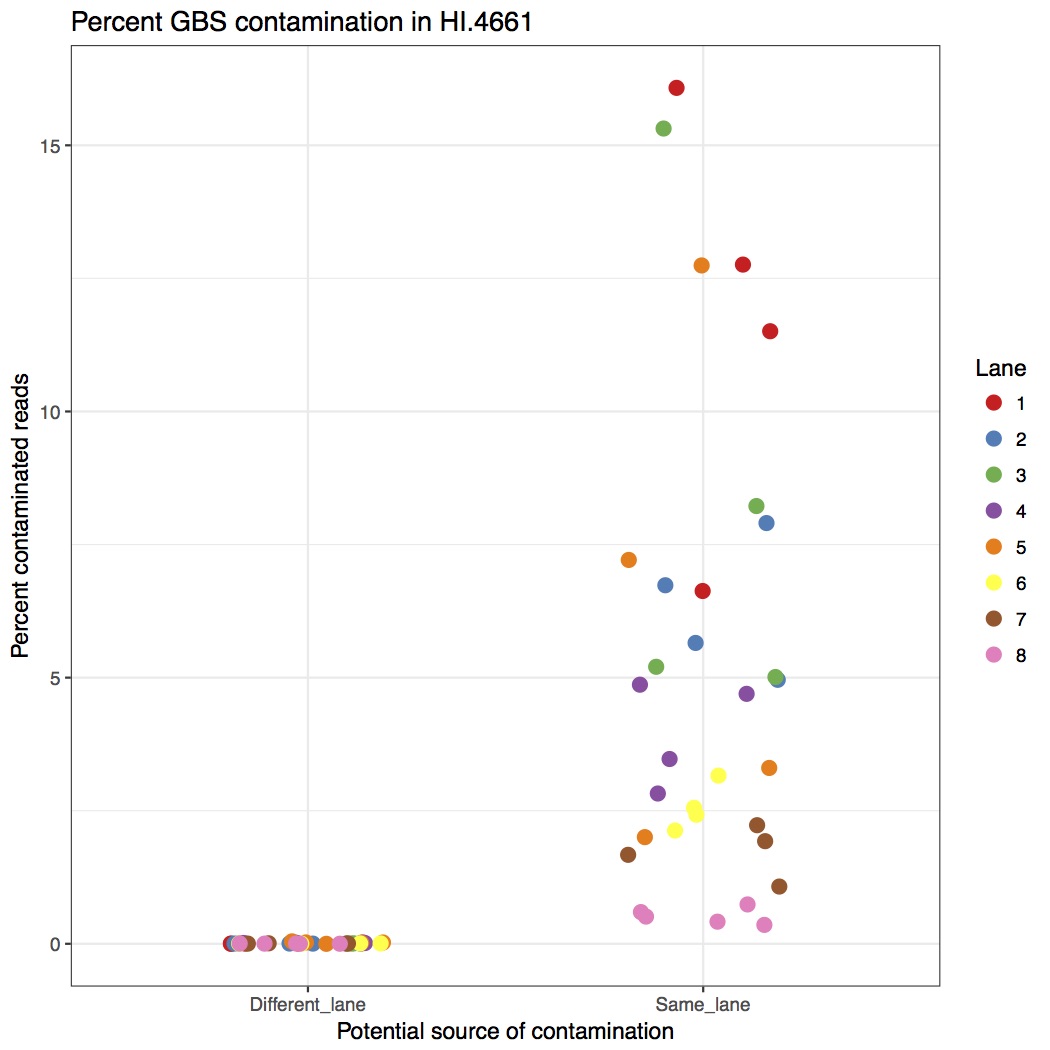
The left side is contamination from barcodes sequenced in different lanes (i.e. ones where they couldn’t contaminate). The right side is barcodes from GBS samples sharing the same lane (i.e. ones that could contaminate. The take home message is that between 1% to 15% of reads are GBS contamination. This is far above what we want to accept so they should be removed.
I’ve written a script to purge barcoded reads from samples. You give it the set of possible barcodes, both forward and reverse (All current barcodes listed here: GBS_2enzyme_barcodes). I’ve been conservative and been giving it all possible barcodes, but you could also trim it to only the barcodes that would be present in the lane. It looks for reads that start with the barcode (or any sequence 1bp away from the barcode to account for sequencing error) plus the cut site. If it finds a barcoded read, it removes both directions of reads. It outputs some stats at the end to STDERR. Note, this was written for 2-enzyme PstI-MspI GBS, although could be rewritten for other combinations.
An example of how you could apply this:
Make a bash script to run the perl script:
input=$1
perl ../purge_GBS_contamination.pl /home/gowens/bin/GBS_2enzyme_barcodes.txt ${input}_R1.fastq.gz ${input}_R2.fastq.gz ${input}.tmp;
gzip ${input}.tmp_R*.fastq
Run this bash script using gnu parallel
ls | grep .gz$ | grep R1 | sed s/_R1.fastq.gz//g | parallel -j 10 bash .decontaminate.sh 2>> decontamination_log.txt