
I’ve often wondered if the TEs in the sunflower genome were producing erroneous SNPs. I have unintentionally produced a little test of this. When calling SNPs for 343 H. annuus WGS samples I set FreeBayes to only call variants in non-TE parts of the genome (as per our TE annotations). Unfortunately, I coded those regions wrong, and actually called about 50% TE regions and 50% non-TE regions. That first set was practically unfiltered, although only SNPs were kept. I then filtered that set to select high quality SNPs (“AB > 0.4 & AB < 0.6 & MQM > 40 & AC > 2 & AN > 343 genotypes filtered with: DP > 2”). For each of these, I then went back in and removed SNPs from TE regions, and I’ve plotted totals below:
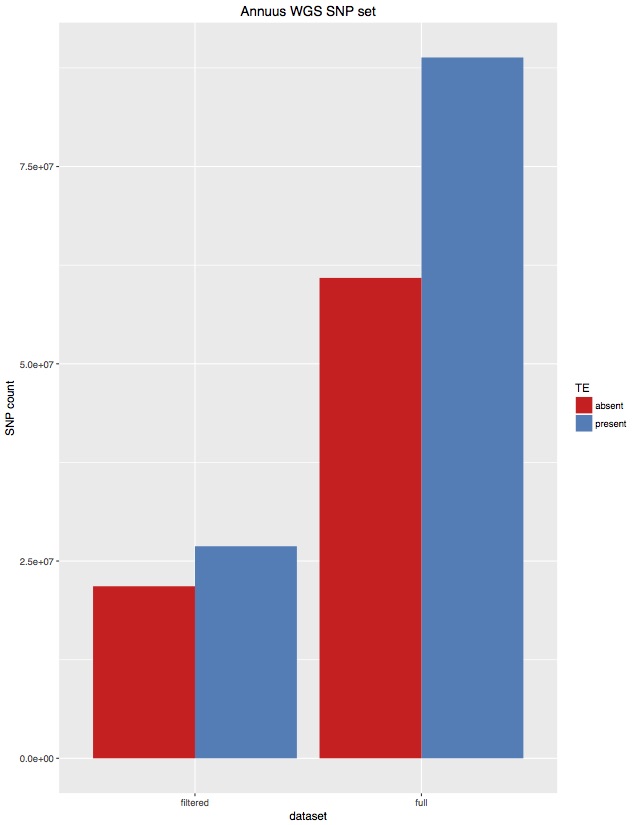
There are a few things we can take from this:
- There are fewer SNPs in TE regions than non-TE regions. Even though the original set was about 50/50, about 70% of SNPs were from the non-TE regions.
- For high quality SNPs, this bias is increased. 80% of the filtered SNPs were from non-TE regions.
Overall, I think this suggests that the plan to only call SNPs in non-TE regions is supported by the data. This has the advantage of reducing the genome from 3.5GB to 1.1GB, saving a bunch of computing time.