When analyzing genomic data, we first need to align to the genome. There are a lot of possible choices in this, including BWA (medium choice), stampy (very accurate) and bowtie2 (very fast). Recently a new aligner came out, NextGenMap. It claims to be both faster and deal with divergent read data better than other methods.
I took a paired read whole genome shotgun sample from H. anomalus prepared by Marco and compared it using different aligners. The file had 31610488 read pairs, that have been trimmed for base quality using trimmomatic. I aligned it to the HA412 bronze genome. H. anomalus is fairly divergent from H. annuus, so this is a challenge. I used the default options for each aligner. Here are the results
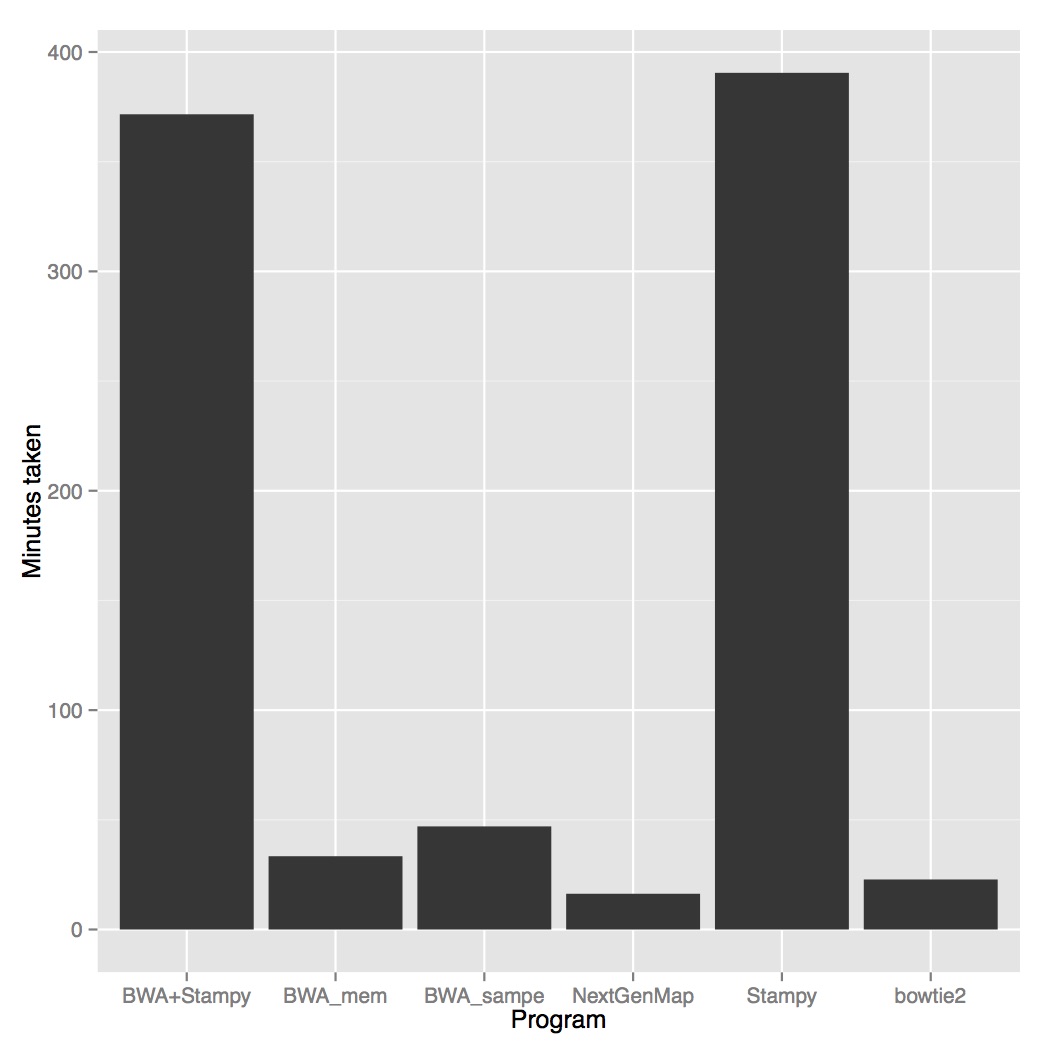
Firstly, NextGenMap was the fastest aligner by far. Bowtie2 was next fastest (1.4X more time), then BWA mem (2X more time), BWA sampe (3Xmore time) and then stampy and stampy+bwa mem (23X more time). NextGenMap also has a fairly slow initial reference loading step, so on larger samples the gains may be even better.
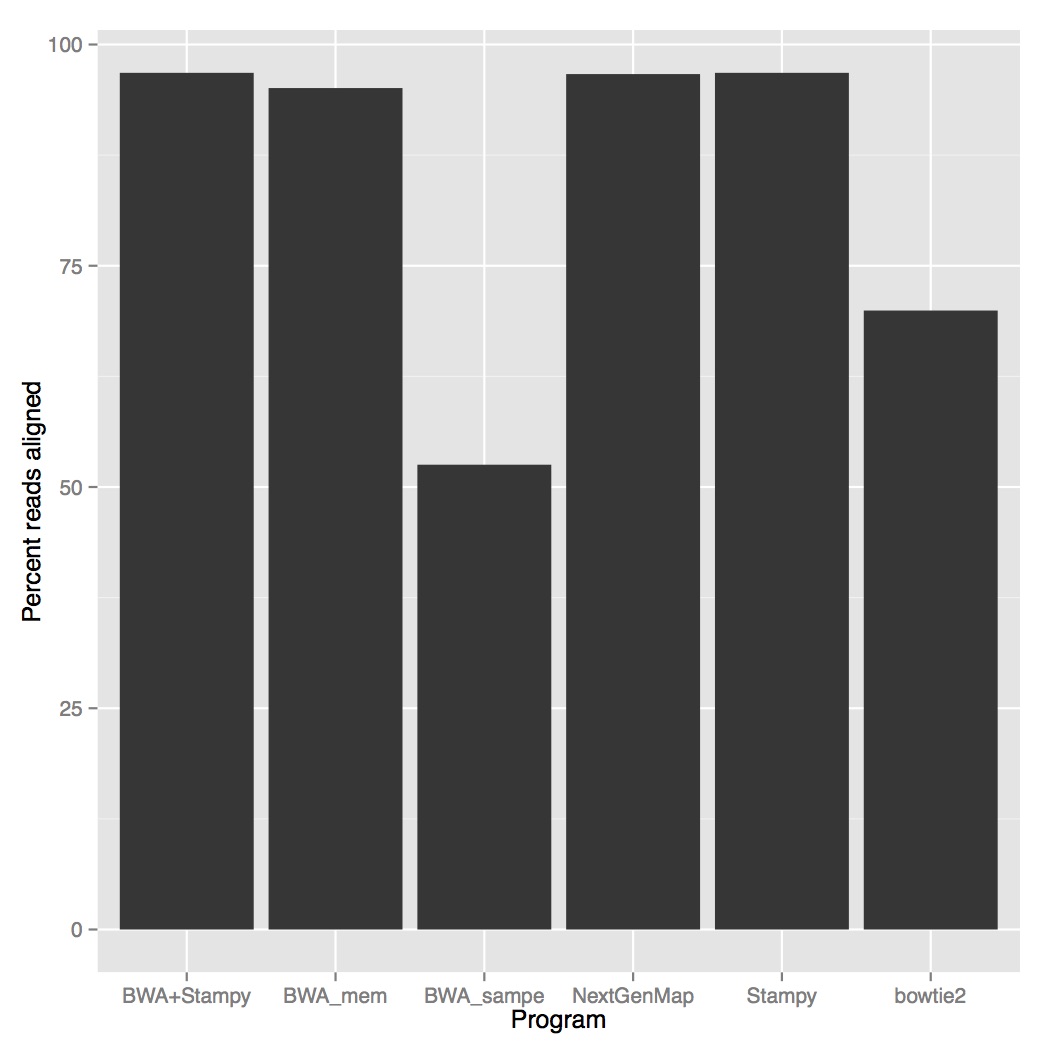
When we look at accuracy, stampy and stampy+bwa were the most accurate (96.8% reads aligned), NextGenMap was slightly lower (96.66%) followed by BWA mem (95.08%). Both BWA sampe (52.52%) and bowtie2 (69.94%) didn’t do very well.
With this being said, just percent reads aligned doesn’t encompass the entire quality of an alignment, but I think this is pretty good evidence that we should be shifting to NextGenMap. It is free, open source, (in this case) the fastest option and pretty much as accurate as the incredibly slow Stampy. Just for reference, I aligned all of Marco’s anomalus WGS on my own linux box over a weekend, which would have taken more than two months with stampy and gotten about the same results.
Here is a software package specifically designed to test aligners. It simulates data based on your reference and checks speed and mapping quality.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4618857/
I haven’t run it, but based on their wild species work they find BWA-mem and NGM have very similar mapping accuracy but NGM is faster. So that’s consistent with my test case.