Hi all,
Here’s another method for DNA extraction. The blog is stuffed to the gills with DNA extraction methods. The current standards are ‘Qiagen-like’ columnless – used to generate the DNA for the genome sequencing project and CTAB. I add this protocol because itis
easy and it is effective.
I extracted from Ha89 and harvested ~ 115 ng/uL from 20 cm tall plants. I also purposely ‘took it slow,’ letting tissue thaw after freezing to see PlantDNAzol’s efficacy. It’s efficient.
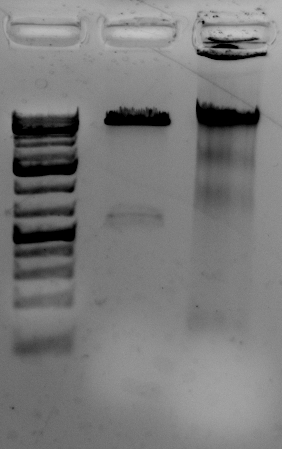
Quality is good
The gel to the left:
Right most lane contains Ha89 genomic DNA – 10 uL loaded of 70 at 115 ng/uL DNA – 260/280 was 1.78. 260/230 was ~1.00 (I suspect I could have added an additional ethanol wash to remove Guandine from the DNA mixture.
Is the DNA useful? Can downstream reactions proceed?
Yes. I digested the DNA with a methylation sensitive restriction enzyme, PstI and a methylation insensitive enzyme, EcoRV
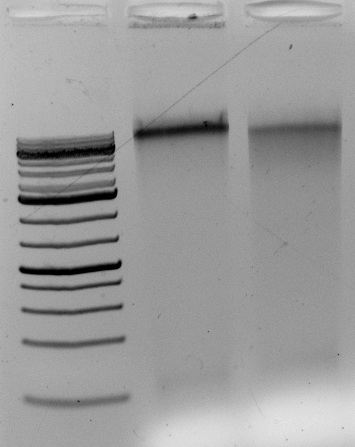
<–The gel to the left :
Leftmost lane Ladder
A vector digested with PstI,
Ha89 gDNA digested with PstI 240 minutes,
Ha89 gDNA digested with EcoRV 240 minutes.
The DNA digests. But does it contain contaminants that upset the enzymes over long incubations? Overnight?
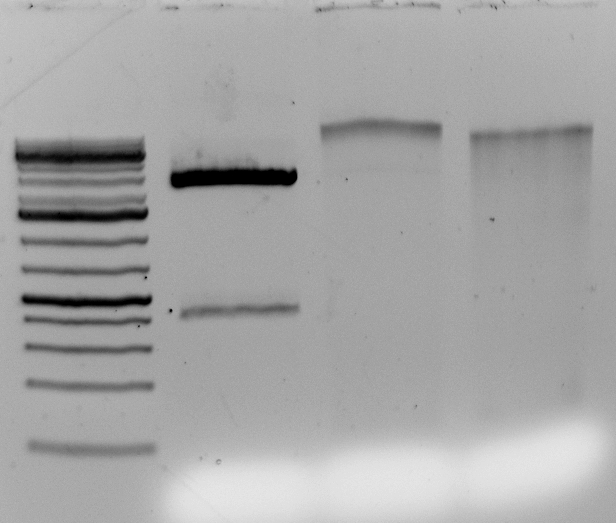
Gel above: From left- Ladder, unrelated vector digest,
Ha89 gDNA digested with PstI 22 hours
Ha89 gDNA digested with EcoRV 22 hours.
Protocol for DNAzol extraction (exactly as published by LifeTech but easier to follow – http://tools.lifetechnologies.com/content/sfs/manuals/10978.pdf:
Have these items on hand:
1 0.6 mL DNAzol per 100 mg sample
2. 0.3 mL chloroform per 100 mg sample
3. Timer
4. 100% ethanol (0.225 mL per sample),
5. 75% ethanol (0.3 mL per sample)
Handle all inversions carefully. When you see invert or shake handle your samples gently
1. Mix 100 mg ground tissue with 0.3 mL PlantDNAzol – 100 mg is max. Overdoing will hurt your yield.
2. Invert gently to aid in lysis and dispersion
3. Once completely dispersed incubate at RT, 5 min, shake periodically.
4. Add 0.3 mL chloroform and mix.
5. Once completely dispersed incubate at RT, 5 min, shake periodically.
6. Centrifuge at RT, 12 000 g (NOT rpm) 10 min
7. Harvest the supernatant. – you’ll see a phenol/chloroform styled triple layer. The middle layer will be pulpy containing your cellulosic debris and proteins. Don’t collect the middle layer. Less is more
8. Mix supernatant from 7 with 225 uL 100% ETOH.
9. Incubate at RT, 5 min
10. Centrifuge mixture 5000 g 4 min – get preparing for step 11
11. Make a PlantDNAzol – Ethanol mixture: For one sample mix 0.3 mL PlantDNAzol with 0.225 mL 100% ethanol.
12. Discard the supernatant from 10 and mix it with 0.3 mL of the mixture prepared in step number 11
13. Incubate as in step 9.
14. Spin as in step 10.
15. Pour off supernatant
16 Wash pellet with 75% ethanol – 0.3 mL – this step can be repeated if your 260/230 isn’t adequate. Guanidine absorbs strongly in 230 nm wavelength
EDIT: repeat step 16 for a total of 2 washes.
17. Spin as in step 10.
18. Remove supernatant – if your samples are green repeat step 16.
19. Resolubilize your DNA in TE or NaOH. Make sure to run your bead of TE over the wall of the tube to collect your DNA.
EDIT – Less is more as is usually the case with DNA extraction. I harvested from 38, 60, and 100 mg of tissue. – A sweet spot for tissue quantity is 45 to 60 mg for the given amount of Plant DNAzol
Qubit quantifiication: 45 mg of tissue yielded 264 ng/uL ug/mL. 60 mg, 305 ng/uL. 100 mg, 12.4 ng/uL




