As you know, the sunflower genome contains a large amount of repetitive sequences, that is why it is so big and so annoying to sequence. I have been working for a while on optimizing a depletion protocol, to try to get rid of some repetitive sequences in NGS libraries (transposons, chloroplast DNA…).
The protocol uses a Duplex Specific Nuclease (DSN), originally isolated in the Kamchatka crab (Risk!), and sold at a rathe hefty price by Evrogen, a Russian company. The idea is to denature and slowly re-annealing your library; fragments from repetitive sequences, being present in higher copy number, will have an easier time finding a suitable complementary strand, and will therefore revert to double-stranded DNA quicker than fragments from single-copy genes. After a while, you stop the re-annealing process and treat the sample with the DSN, which degrades specifically double stranded DNA. Repetitive sequences will therefore be degraded preferentially, reducing their relative abundance in your library.
This is of course not my idea, but I spent some time to (hopefully) improve the protocol. Things I changed with respect to the protocol described in Matvienk et al. 2013 in PLoS ONE, and that in my hands increased depletion efficiency (as quantified by qPCR assays) are:
– Increased initial library concentration (up to 160 ng/micoliter)
– Increased annealing temperature to 78°C
– Increase reaction temperature to 70°C
– Reduced enzyme concentration to 1/10 (also reduces cost of the enzyme to about 50 cent per reaction)
– Reduced reaction time to 15 minutes
– Used libraries with shorter, incomplete adapters (see the end of the blog)
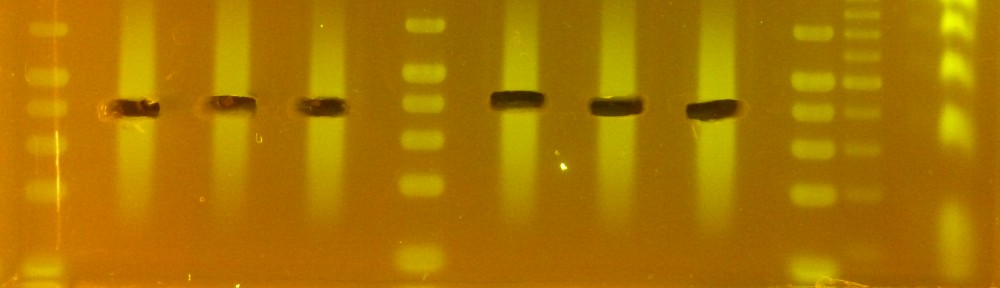
When used on H. anomalus libraries (the following numbers are based on actual sequencing data), in its current form the DSN treatment results in a 4-8 fold decrease in coverage for chloroplast DNA, 20-30 fold decrease in coverage for ribosomal RNA, and about a 3 fold increase in coverage for single copy genes (I used a set of 12 flowering time genes from one of Ben’s manuscripts for that). Determining the precise reduction in transposons abundance is trickier (as it depends a lot on what you manage to align to your reference, and I only had data from H. annuus). In my opinion though the more relevant data is the coverage of single copy genes, which is what you are really looking for when using this protocol. The deduction from those data is that the approximate size of the genome is “reduced” to about 1/3 after DSN treatment. I am sure someone with better bioinformatics skills could make a much better job at estimating the actual depletion efficiency, but that’s as far as I got.
Here is the protocol, and after that a few considerations about adapters and primers.
Depletion of repetitive sequences – WGS libraries
A few considerations about adapters and primers
As I just mentioned, the idea behind the DSN treatments is that repetitive or abundant sequences will anneal more easily and will therefore be preferentially degraded by the DSN. The problem with this is that actually all your fragments are flanked by identical sequences, i.e. the adapters. The longer the adapter, the more likely it is that it will be degraded. You can see that when you try to amplify, after DSN treatment, a regular library with full-length adapters. If you use short PCR primers that anneal at the extremities of the adapters, you’ll get very poor amplification. If instead you use longer primers, that span most of the adapters, you’ll get much better amplification. This is likely because the DSN enzyme cuts inside the adapter, leaving fragments with only partial adapters. In order to be able to amplify (and then sequence) these fragments, you’ll need to use longer PCR primers that will anneal also to partially truncated adapters, and complete them during the following PCR cycles.
Using longer PCR primers therefore solves the problem of partially digested adapters. The region of the adapters that these long PCR primers anneal to includes also the sequence of the index (the barcode embedded in the reverse adapter most current libraries use), and you have therefore to use primers with the same index. That means that you cannot pool your libraries before the DSN treatment.
Still, this also means that when using full-length adapters a lot of the cutting the DSN does is because of adapters annealing, and has nothing to do with the depletion. A partial fix to this is using shorter, incomplete adapters when making your library (as for example the ones in the protocol I posted some time ago), and amplify it before the DSN treatment using PCR primers that do not extend the adapters. This will leave you with a library ready for DSN treatment that has much shorter complementary sequences at the end of each fragments, reducing a-specific cleavage and (apparently) increasing depletion efficiency. Once you are done with the DSN treatment you can complete your adapters (and barcode your samples) during PCR amplification.