
You are required to login to view this page.
Image analysis
You are required to login to view this page.
Tassel 5
Tassel 5.0 is out and include many GBS functions. Although it is GWAS oriented it could still do a lot of preprocessing for other purposes:
- Bit level encoding of nucleotides so genetic distance and linkage disequilibrium estimates can be made very quickly (20-50X speed increases).
- Extensive use the HDF5 file format, which has been developed as a robust element of many climate modelers for matrix style data
- Tools for extracting and calling SNPs from extensive Genotyping-by-Sequencing data (tested for 60,000 samples by over 2.5 million SNPs and 96 million sequence alleles).
- Projection and imputation procedures that are optimized for the large families in crops. Some of these optimizations permit memory and computational improvements of >100,000 fold.
- Mixed models based on DNA relationships have come to dominate GWP (Meuwissen et al 2001) and GWAS (Yu et al 2006), yet these models can be slow to solve. TASSEL has been a test bed and implements some of the most best optimizations, such as EMMA (Kang at al 2008), plus approaches optimize variance components once P3D (Zhang et al 2010) and EMMAX (Kang et al 2010). Compression algorithms are also available (Zhang et al 2010). When used correctly, these optimizations make powerful GWAS computationally possible.
- The code is being continually optimized for larger numbers of cores and clusters. For example, we generally run imputation on 64-core machines. And while Java provides some excellent is interoperability between systems, its code is about 2-fold slower than optimized C libraries, and 10-fold slower than GPU processing for some problems. TASSEL5 is building out connection layers directly to native code, when these efficiencies are need.
PstI-MspI GBS: an update
The good news: it looks like PstI-MspI GBS offers substantial benefits over PstI alone, even without depleting high copy fragments. Marco’s DSN protocol may improve things further, and we have a trial sequencing library running now.
Depletion of repetitive sequences – WGS libraries
As you know, the sunflower genome contains a large amount of repetitive sequences, that is why it is so big and so annoying to sequence. I have been working for a while on optimizing a depletion protocol, to try to get rid of some repetitive sequences in NGS libraries (transposons, chloroplast DNA…). Continue reading
Non-Batch Submissions to the SRA
Since many of you have smaller amounts of data (a handful of samples/runs per experiment), I wanted to provide some info on submitting that data to the SRA without the help of their staff. The SRA has recently re-vamped their submission process, and it is much easier/simpler than before.
It’s important that everyone submit their own data. The SRA provides a free off-site backup, and we have already replaced corrupted files on our computers with clean copies obtained from the SRA. The time you spend doing this will ensure that you always have a clean copy of your raw data.
Here is a brief overview of the submission process, as well as the info you’ll need.
As well as the NCBI’s “quick start guide”.
Here are the pages for submitting BioProjects and BioSamples:
The process now is fairly straightforward and well-documented by the SRA. If anyone has trouble, you can ask others in the lab that have submitted their own data in the last year or so. I believe Dan Bock is one of them.
Here are some of the lab’s existing bioprojects. These can give you an idea of what kind of info to include in the abstract/project description, etc.:
http://www.ncbi.nlm.nih.gov/bioproject/PRJNA64989
http://www.ncbi.nlm.nih.gov/bioproject/PRJNA194568
http://www.ncbi.nlm.nih.gov/bioproject/PRJNA194569
http://www.ncbi.nlm.nih.gov/bioproject/PRJNA194570
http://www.ncbi.nlm.nih.gov/bioproject/PRJNA194446
http://www.ncbi.nlm.nih.gov/bioproject/PRJNA194445
The limits of GBS sample size
Image
I’ve been doing work on a stickleback GBS dataset and we’re trying to figure out how many samples we can cram into a single lane of illumina. I did some analyses which people may find useful. It’s unclear how applicable the recommendations are for sunflower which seems to have more problems than fish.
Take home message, with 25% less data you lose around 15% of the genotype calls, but up to 50% of the SNPs if you use a stringent coverage filter, due to how the lost data is distributed among loci and individuals.
Mounting the Moonrise NFS
Edit: As of February 2015, all our servers are running CentOS 7. The original instructions below are for Debian/Ubuntu linux, but here is a better set of generalized instructions for CentOS: https://www.howtoforge.com/nfs-server-and-client-on-centos-7
If you are mounting on an Ubuntu/Debian system, you can still use the instructions below. If you are mounting on a Red Hat derivative (Fedora, RHEL, CentOS, ScientificLinux, etc.), the link above should work.
I just had to re-learn how to do this today, so I thought it would be a good idea to write it up.
If any of you would like to mount the NFS on a computer (Unix, and static IPs only. This means no wireless!) in the building, you can do so at your convenience using this guide.
First, install nfs-common with your package manager (ubuntu: apt-get install nfs-common)
Next, create a folder for the mount to bind to on your computer, and make sure the permissions are set to 777:
user@mycomputer: mkdir -p /nameofyourNFSfolder
user@mycomputer: chmod 777 /nameofyourNFSfolder
I think the whole tree needs the same permissions, so what I’ve done for all our machines (and what seems easiest) is to make a folder in the root directory, so that you don’t have to worry about the permissions in parent folders.
Next, the /etc/hosts.allowed and /etc/exports files on moonrise need to be modified. Chris, Frances, and I all have the necessary permissions to do this. Just ask one of us and we can do it.
root@mooonrise: nano /etc/exports
Add the following to the line beginning with /data/raid5part1
137.82.4.XXX(rw,sync) (with XXX standing in for the static IP of your machine)
You could also do this with machines in other buildings/off-campus as long as their IPs are static.
root@moonrise: nano /etc/hosts.allow
Your IP has to be added to the end of each line in this file.
Now reload the /etc/exports file on moonrise (a full NFS restart is not required, and will unmount it on other machines! don’t do that unless you know for sure that no one is using the NFS on any of our computers!)
root@moonrise: exportfs -a
Finally, mount the NFS on your machine:
user@mycomputer: sudo mount -v -o nolock -t nfs moonrise.zoology.ubc.ca:/data/raid5part1 /nameofyourNFSfolder
There are various options you can use with the mount command, but the above should work for just about anyone.
If you want it to auto-mount each time you boot your computer, you can add the following lines to your /etc/fstab file:
#moonriseNFS
137.82.4.123:/data/raid5part1 /nameofyourNFSfolder nfs auto 0 0
That’s it!
Quick update: 96-well plate CTAB DNA extraction with fresh tissue
When I posted the protocol I have been using for CTAB DNA extraction in 96-well plates, I included results from a few plates I did starting from dried H. anomalus leaves I collected a couple of months earlier in Utah. While they showed that the method worked well enough when starting from “difficult” material, they were not exactly what you’d dream of when you decide to extract DNA, especially if you are starting instead from fresh material.
Here are the results from a plate of extraction I did starting from individual small (1.5-2 cm in length), young leaves from ~3 month-old H. anomalus plants. I collected the leaves directly in 96-well plates (I already put one metal bead in each well), put them on dry ice until I got to the lab (a couple of hours), left them overnight in the -80, and started extracting DNA the day after.
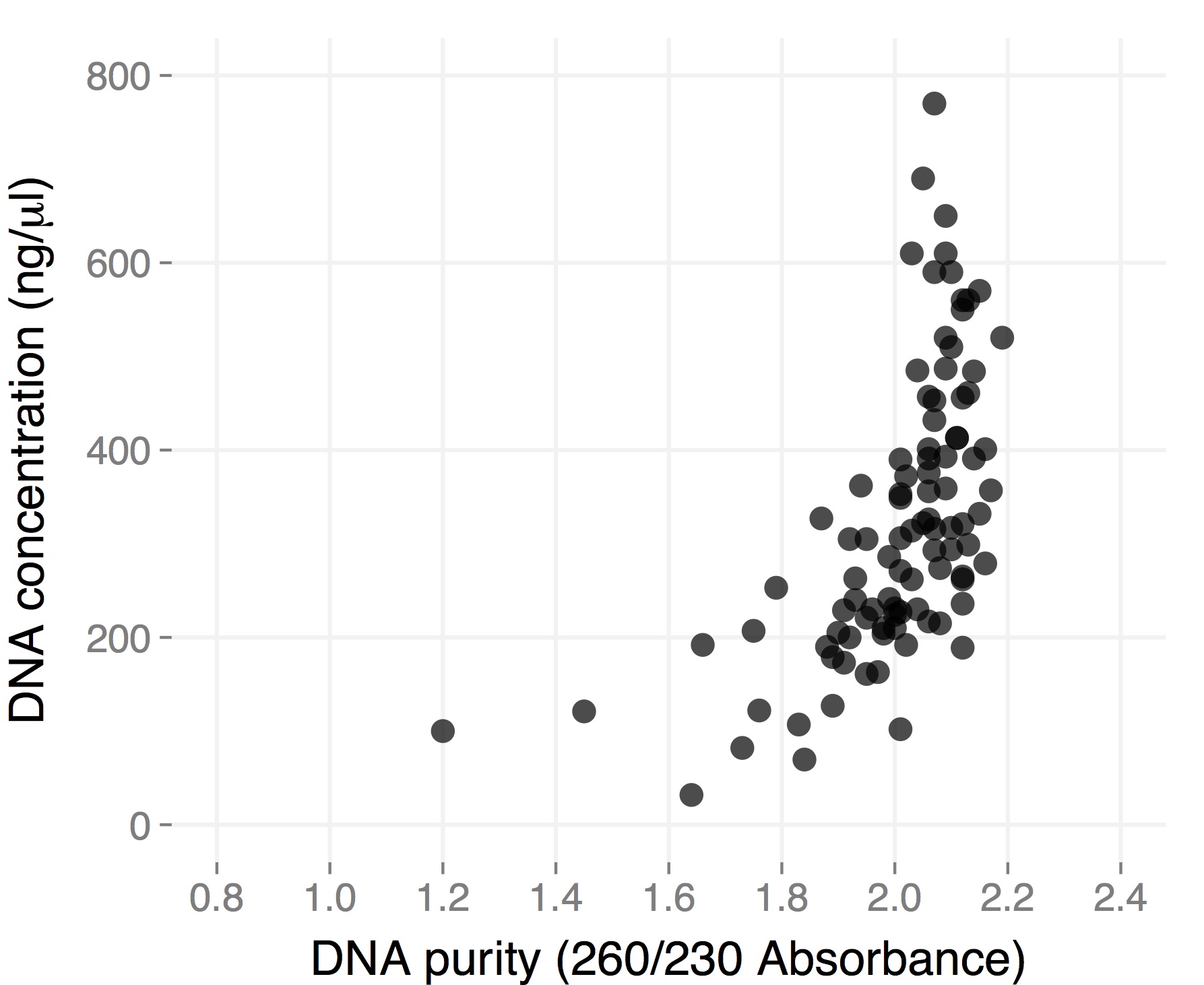
The final volume was 50 microliters, so total yield is for most samples between 10 and 30 micrograms of DNA. These are “real” DNA concentration measured by Qubit. Both average yield and purity are considerably higher than for dry tissue, and they are comparable to what you would get starting with frozen tissue using the single tube protocol (but you save a ton of time). Hope this gets you all more thrilled about 96-well plate DNA extractions 🙂
SNP calling with GATK warning
I’ve come to the realization that I’ve been making a mistake in my SNP calling using GATK. I’ve been filtering for site quality not for individual genotype quality. This is a critical difference and leads to some dubious SNPs being called.
It works like this. For each position in the genome, GATK will use all the data together to call whether an alternate allele exists. This is represented by the phred scaled QUAL score. A high QUAL score means that the alternate allele exists in in your dataset. This number is often very high if you’re analyzing a whole plate of GBS because it takes into account all the data from all the individuals. I’ve been filtering based on this number.
There is also a MQ score, which is the mapping quality for all the reads at that site.
For each individual, if there are any reads at that site GATK will output a genotype call (i.e. 0/0, 0/1, 1/1). It also outputs the number of reads supporting each allele, the QT score (which is the phred scaled probability of that the genotype call being correct) and the likelihoods of each possible genotype. When you use select variants in GATK, it filters sites but not genotypes (so you can have a site that is good, but individual genotype calls at that site that are bad).
If you use vcf-to-tab to convert your vcf to tab separated it will accept every genotype call. This means that for some individuals you will have 1 read supporting the reference and it will be called as 0/0. The QT score will be 3, which is incredibly low, but vcf-to-tab ignores that information and calls it. Therefore a proportion of your SNPs are highly questionable. This likely contributes to the heterozygote loss seen in GBS data.
Sam and Chris wrote a script that converts a vcf file to tab but also makes sure that the quality (QUAL, QT, MQ, and depth) is good. I’ll post it if they’re OK with it be up.
Edit: Here are the scripts. They are identical except one outputs in IUPAC format (AT=W, AC=M, etc) and the other outputs in the standard double nucleotide format (AA, AT, etc).vcf2vertical_dep_GATK-UG vcf2vertical_dep_GATK
Also note that sometimes GATK will output a genotype call that looks like:
./.:.:3
While normally they should look like:
./. or 0/0:17,0:18:45:0,45,353
This is saying that there are three reads at this position, but the quality is so bad that GATK is not calling the genotype. If you have these, the scripts will throw an error but will correctly output an NN for that genotype. So if you get a normal output and many errors, it’s probably OK.
BayEnv
Apparently BayEnv is the way to do differentiation scans. Here I offer some scripts and advice to help the you use Bayenv.
Install, no problem get it here: http://gcbias.org/bayenv/ Unzip.
I have to say the documentation is not very good.
There is 2 things you will probably want to use BayEnv for:
1) Environmental correlations
2) XtX population differentiation
I will just be showing XtX here. Both need a covariance matrix. This is not the “matrix.out” file you get from the matrix estimation step.
. But first the SNP table.
Here is a script Greg O made to make a “SNPSFILE”* for bayenv from our standard SNP table format: SNPtable2bayenv I’ve made a small modification so that it excludes any non-bi-allelic site and to also print out a loci info file.
Use it like this:
perl SNPtable2bayenv.pl SNPfile POPfile SampleFile
This will make three files with “SampleFile” appended to the start of the name.
*not actually a SNP file at all, it is an allele count file.
Input SNPfile (all spaces are tabs):
CHROM POS S1 S2 S3 S4 S5 chr1 378627 AA TT NN TT AA chr2 378661 AA AG NN GG AA chr3 378746 GG AA NN AA GG chr4 383246 NN GG NN GG TT chr5 397421 CC GG NN GG CC chr6 397424 GG AA NN AA GG chr7 409691 NN CC NN NN GG chr8 547653 AA GG NN GG AA chr9 608412 AA GG NN NN AA chr10 756412 GG CC NN CC GG chr11 1065242 NN TT NN NN CC chr12 1107672 NN TT NN TT NN
POPfile (all spaces are tabs):
S1 p1 S2 p1 S3 p2 S4 p2 ...
Now estimate the covarience matrix:
./bayenv2 -i SampleFile.bayenv.txt -p 2 -k 100000 -r21345 > matrix.out
Now this is “matrix.out” file is not what you actually want. You have to use it to make a new file that contains a single covariance matrix. You could use “tail -n3 matrix.out | head -n2 > CovMatrix” or just cut it out yourself. Currently I am just using the last (and hopefully converged) values.
My CovMatrix.txt looks like this (again single tabs between):
3.009807e-05 -1.147699e-07 -1.147699e-07 2.981001e-05
You will also need a environment file. I am not doing the association analysis so am just using some fake data. It is one column for each population and each row is the environmental values. Tab separated.
FakeEnv.txt
1 2
Now you can do the population differentiation test for each snp using a “SNPFILE”. I bet you think you are ready to go? Not so fast, you need a “SNPFILE” not a “SNPSFILE”. Not that either actually really have SNPs in them, just allele counts. Not that I am bitter. SNPFILE is just one site from the SNPSFILE. Here is a script that takes your “SNPSFILE” and runs the XtX program on each of the sites: RunBayenvXtX
perl RunBayenvXtX.pl SampleFile.bayenv.txt SampleFile.LocInfo.txt CovMatrix.txt FakeEnv.txt 2 1
Where 2 is the number of populations and 1 is the number of environmental variables.
This will produce a file that has the site, allelecountfile name and the XtX value.
FTP to a server
If you want to download something from a sequence read archive, often the files are available on ftp. A browser will automatically download those ftp files if you click the link, but if you are working on a server, it is a pain to download the file to your home computer and then upload it. Here is a quick guide to do this on the command line.
1)Copy the link address for the ftp file you want, from the website.
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR168/ERR168678/ERR168678_2.fastq.gz
2) SSH to your server and navigate to the folder your want to put the data in.
3) Type ‘ftp’. You are now in the ftp prompt.
ftp
4) Open a connection to the ftp server by typing in ‘open <server name>’. It asks you for a name, and if you don’t have one try ‘anonymous’. It asks for a password, try just pressing enter.
ftp> open ftp.sra.ebi.ac.uk
Connected to ftp.sra.ebi.ac.uk.
220-
220- ftp.era.ebi.ac.uk FTP server
220
Name (ftp.sra.ebi.ac.uk:owens): anonymous
331 Please specify the password.
Password:
230 Login successful.
Remote system type is UNIX.
Using binary mode to transfer files.
5) Navigate to the file you want using ‘cd’ to change directory and ‘ls’ to view directory contents.
ftp> cd vol1/fastq/ERR168/ERR168678
250 Directory successfully changed.
ftp> ls
200 PORT command successful. Consider using PASV.
150 Here comes the directory listing.
-r–r–r– 1 ftp ftp 10990053136 Dec 22 2012 ERR168678_1.fastq.gz
-r–r–r– 1 ftp ftp 10998002687 Dec 22 2012 ERR168678_2.fastq.gz
226 Directory send OK.
6) Download data using ‘get <filename>’
get ERR168678_2.fastq.gz
7) Your data is now downloading. Its best to run this in a screen so that you don’t have to keep the window open, in case it runs slowly.
Approximate Bayesian Computations
In many cases it may be more straightforward (and informative) to test specific models using our data. An interesting approach for inferring population parameters and/or model testing is approximate Bayesian computations (ABC). There are several available tools such as msBayes, DIYABC, PopABC abctools R package, ABCTools.
Although ABC is a powerful and useful approach it has some caveats, e.g. choice of summary statistics, number and complexity of the models tested, amount of data and more. For realistic expectations and simple models ABC could really add some interesting insights to popgen studies.
Paired End for Stacks and UNEAK
Both stacks and uneak are made for single end reads. If you have paired end data here is a little cheat that puts “fake” barcodes onto the mate pairs and prints them all out to one file. It also adds the corresponding fake quality scores.
perl GBS_fastq_RE-multiplexer_v1.pl BarcodeFile R1.fastq R2.fastq Re-barcoded.fastq
BarcodeFile should look like (same as for my demultiplexing script) spaces must be tabs:
sample1 ATCAC
sample2 TGCT
…
# note this could also look like this:
ATCAG ATCAG
TGCT TGCT
…
As it does not actually use the names (it just looks at the second column).
Here it is:
GBS_fastq_RE-multiplexer_v1
actual UNEAK
Some of you may have heard me ramble about my little in house Uneak-like SNP calling approach. I am being converted to using the actual UNEAK pipeline. Why? reason #1 is good god is it fast! I processed 6 lanes raw fastq to snp table in ~1 hour. #2 I am still working on – this will be a comparison of SNP calls between a few methods but I have a good feeling about UNEAK right now.
Here is the UNEAK documentation: http://www.maizegenetics.net/images/stories/bioinformatics/TASSEL/uneak_pipleline_documentation.pdf
It is a little touchy to get it working so I thought I would post so you can avoid these problems.
First install tassel3 (not tassel 4) you will need java 6 or younger to support it: http://www.maizegenetics.net/index.php?option=com_content&task=view&id=89&Itemid=119
To be a hacker turn up the max RAM allocation. Edit run_pipeline.pl to change the line “my $java_mem_max_default = “–Xmx1536m”;” to read something like “my $java_mem_max_default = “–Xmx4g”;” (That is 4g for 4G of RAM).
Go to where you want to do the analysis. This should probably be a new and empty directory. Uneak starts by crawling around looking for fastq files so if the directory has some files you don’t want it using it is going to be unhappy.
Make the directory structure:
../bin/run_pipeline.pl -fork1 -UCreatWorkingDirPlugin -w /home/greg/project/UNEAK/ -endPlugin -runfork1
Move your raw (read: not demultiplexed) fastq or qseq to /Illumina/. If you are using fastq files the names need to look like this: 74PEWAAXX_2_fastq.txt NOT like this:74PAWAAXX_2.fastq and probably not a bunch of other ways. It has to be flow cell name underscore lane number understore fastq dot txt. You might find others work but I know this does (and others don’t).
Make a key file. This is as described in the documentation. It does not have to be complete (every location on the plate) or sorted. Put it in /key/
Flowcell Lane Barcode Sample PlateName Row Column 74PEWAAXX 1 CTCGCAAG 425218 MyPlate7 A 1 74PEWAAXX 1 TCCGAAG 425230 MyPlate7 B 1 74PEWAAXX 1 TTCAGA 425242 MyPlate7 C 1 74PEWAAXX 1 ATGATG 425254 MyPlate7 D 1 ...
Run it. Barely enough time for coffee.
../bin/tassel3.0_standalone/run_pipeline.pl -fork1 -UFastqToTagCountPlugin -w /home/greg/project/UNEAK/ -e PstI -endPlugin -runfork1 # -c how many times you need to see a tag for it to be included in the network analysis ../bin/tassel3.0_standalone/run_pipeline.pl -fork1 -UMergeTaxaTagCountPlugin -w /home/greg/project/UNEAK/ -c 5 -endPlugin -runfork1 # -e is the "error tolerance rate" although it is not clear to me how this stat is generated ../bin/tassel3.0_standalone/run_pipeline.pl -fork1 -UTagCountToTagPairPlugin -w /home/greg/project/UNEAK/ -e 0.03 -endPlugin -runfork1 ../bin/tassel3.0_standalone/run_pipeline.pl -fork1 -UTagPairToTBTPlugin -w /home/greg/project/UNEAK/ -endPlugin -runfork1 ../bin/tassel3.0_standalone/run_pipeline.pl -fork1 -UTBTToMapInfoPlugin -w /home/greg/project/UNEAK/ -endPlugin -runfork1 #choose minor and max allele freq ../bin/tassel3.0_standalone/run_pipeline.pl -fork1 -UMapInfoToHapMapPlugin -w /home/greg/project/UNEAK/ -mnMAF 0.01 -mxMAF 0.5 -mnC 0 -mxC 1 -endPlugin -runfork1
Some bioinformatics tools
New tools for analyzing NGS are coming out occasionally. Usually we tend to stick to the same tools just because they work. Well, you may find new tools faster or more accurate for your purposes. Here is a list of tools (and links) which could be used for different purposes (not only RNA-Seq). I could recommend FastQC, FastX, and trimmomatic for cleaning any raw data, and sabre is a nice tool to clean GBS.
FLASH works well for merging reads (there are more tools though) and this seems to be an interesting pre-processing approach. As for sequence aligners, except for bowtie and bwa you may find subread to be fast due to it’s different approach and additional complementary tools for splice alignments, feature counting and SNP calling. Another interesting aligner is stampy which enable to align reads to a diverged reference genome but need some pre- and post- processing to get fast results, otherwise it takes forever.
Of course, there are also some more ‘traditional’ tools for aligning bigger sequences such as MUMer (nucmer, promer) and AMOS. Although these tools are powerful they need some extra pre/post processing which is not always appreciated.
fastSTRUCTURE is fast.
fastSTRUCTURE is 2 orders of magnitude faster than structure. For everyone who has waited for structure to run this should make you happy.
Preprint of the manuscript describing it here: http://biorxiv.org/content/early/2013/12/02/001073
UPDATE comparison: Structure_v_FastStructure Structure (top) 8 days and counting FastStructure (below) 15 minutes. You can see the are nearly identical. I would expect them to be within the amount of variation you normally see on structure runs. Structure is only on K=4 (with 50,000 burnin and 100,000 reps and 5 reps of each K) where FastStructure took just under 15 minutes to go K of 1 to 10. Note: these are sorted by Q values not by individuals so, although I think it is highly unlikely, they may be discordant on an individual scale.
UPDATE on speed: ~400 individuals by ~4000 SNPs K=1 to K=10 in just under 15 minutes.
UPDATE on usage: Your structure input file must be labeled .str but in when you call the program leave the .str off. If you don’t adds it itself and will look for MySamples.str.str
It has a few dependencies which can make the installation a little intimidating. I found installing them was relatively easy and I downloaded everything, installed and tested it all in less than one hour.
The site to get fastSTRUCTURE:
http://rajanil.github.io/fastStructure/
You can follow along there, the instructions are pretty good. This is just what I did with a few notes.
At the top you can follow links to the dependencies.
Scipy + Cython
#Scipy sudo apt-get install python-scipy python-sympy #Cython download + scp tar -zxvf Cython-0.20.tar.gz cd Cython-0.20/ make sudo python setup.py install
Numpy
#probably the easiest way: sudo apt-get install python-numpy python-nose #what I did before I figured out above: download + scp tar -zxvf numpy-1.8.0.tar.gz cd numpy-1.8.0 python setup.py build --fcompiler=gnu #also installed "nose" similarly it is at least needed to run the test #test python -c 'import numpy; numpy.test()'
On to fastSTRUCTURE
wget --no-check-certificate https://github.com/rajanil/fastStructure/archive/master.tar.gz #then #check where GSL is ls /usr/local/lib # You should see a bunch of stuff starting with libgsl # You can look in your LD_LIBRARY_PATH before adding that path echo $LD_LIBRARY_PATH # add it export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/usr/local/lib # check it worked echo $LD_LIBRARY_PATH #mine only has ":/usr/local/lib" in it #decompress the package tar -zxvf master.tar.gz cd fastStructure-master/vars/ python setup.py build_ext --inplace cd .. python setup.py build_ext --inplace #test python structure.py -K 3 --input=test/testdata --output=testoutput_simple --full --seed=100
Where to submit your manuscript?
I was recently made aware of an online tools called JANE (Journal/ Author Name Estimator), which helps you to decide where you should submit a recent paper you are working on. Essentially you copy and paste your abstract into the online tool and it spits out a ranked list of journals where you should submit your paper. It seems to work pretty well. If you test it with abstract of recently published papers, the journal where the paper is actually published almost always comes up in the top 3 choices. JANE also has other functionality of potential use, like finding citations or authors related to your abstract.
This is where it gets interesting.
I read a little more about how the program works. It basically pulls all published abstracts from pubmed and then text mines them for keywords found in your abstract. Now say you’ve written a manuscript; you run it through JANE and it tells you that the best fit is “American Journal of Botany”. But secretly, you hoped that your manuscript would go into a higher profile journal like say “Genetics”. Should you give up all hopes and submit it to AJB? Of course not! What you should do is go look at abstracts published in Genetics and AJB and find out what are the key differences between them. Often, they are surprisingly subtle and by making slight modifications to your abstract, all of a sudden it can become a great fit for Genetics!
So this online tool can actually be very useful to write abstract in the style of the journal you wish to submit to. In addition, I know at least one senior editor at a high profile journal who uses this tool to guide decisions to send manuscript out for review. This is probably not a good decision on their part, but at least now you can use it to your advantage!
GATK on sciborg
A few notes on GATK.
1. GATK requires a younger version of Java than what is on the cluster currently.
> java -jar ./bin/GenomeAnalysisTK-2.8-1-g932cd3a/GenomeAnalysisTK.jar --help Exception in thread "main" java.lang. UnsupportedClassVersionError: org/broadinstitute/sting/gatk/ CommandLineGATK : Unsupported major.minor version 51.0
Get the linux 64 bit version here:
http://java.com/en/download/
move it to sciborg:
> scp ../Downloads/jdk-7u45-linux-x64.tar.gz user@zoology.ubc.ca:cluster/bin/ #ssh to the cluster extract it: > tar -zxvf jdk-7u45-linux-x64.tar.gz #Add it to your path file or use it like so: > ./bin/jdk1.7.0_45/bin/java -jar ./bin/GenomeAnalysisTK-2.8-1-g932cd3a/GenomeAnalysisTK.jar --help
2. You must have a reference with a relatively small number of contigs/scaffolds
Kay identified and addressed this problem and wrote this script: CombineScaffoldForGATK. I’ve modified it very slightly.
perl CombineScaffoldForGATK.pl GenomeWithManyScaffolds.fa tmp.fa
WARNING: this can print empty lines. The script could be modified to address this but I don’t want to break it. Instead you can fix it with a sed one liner:
sed '/^$/d' tmp.fa > GenomeWith1000Scaffolds.fa
3. You must also prepare the “fasta file”
You also have to index it for BWA with bwa index but GATK needs more, see: http://gatkforums.broadinstitute.org/discussion/1601/how-can-i-prepare-a-fasta-file-to-use-as-reference
In short (in the same dir as your genome):
>java -jar ../bin/picard-tools-1.105/CreateSequenceDictionary.jar R= Nov22k22.split.pheudoScf.fa O= Nov22k22.split.pheudoScf.dict >samtools faidx Nov22k22.split.pheudoScf.fa
Where does all the GBS data go? Pt. 2
An analysis aimed at addressing some questions generated following discussion of a previous post on GBS…
Number of fragments produced from a ‘digital digestion’ of the Nov22k22 Sunflower assembly:
Clai: 337,793
EcoRI: 449,770
SacI: 242,163
EcoT22I: 528,709
PstI: 129,993
SalI: 1,210,000
HpaII/MspI: 2,755,916
Here is the size distribution of those fragments (omitting fragments of >5000bp):
All the enzymes
With Msp removed for clarity
Take home message: PstI produces fewer fragments of an appropriate size than other enzymes. It looks like the correlation between total number of fragments and number that are the correct size is probably pretty high.
Now for a double digestion. Pst and Msp fragment sizes and again omitting fragments >5000bp. This looks good. In total fragment numbers (also omitting >5000bp fragments):
Pst+Msp total fragments: 187271
Pst+Msp 500<>300bp: 28930
Pst alone total: 79049
Pst alone 500<>300bp:6815
Take home: Two enzyme digestion could work really well. It may yield more than 4 times more usable fragments. I do think we could aim to get even more sites. Maybe some other RE combination could get us to the 100,000 range. With a couple of million reads per sample this could still yield (in an ideal world) 10x at each site. Send me more enzymes sequences and I can do more of the same.
Edited for clarity and changed the post name