There is now a second set of barcoded adapters that allows higher multiplexing. They also appear to address the quality issues which have been observed in the second read of GBS runs.
This blog post has 1) Info on how to use the barcodes and where they are and 2) some data that might convince you to use them.
Usage
These add a second barcode to the start of the second read before the MSP RE site. The first bases of the second read contain the barcode, just like with the first read. Marco T. designed and ordered these and the info needed to order them is here: https://docs.google.com/spreadsheets/d/1ZXuHKfaR1BYPBX6g0p9GdZHp_21A3z_9pPt_aW0amwM/edit?usp=sharing
I’ve labeled them MTC1-12 and the barcode sequences are as follows.
MTC1 AACT
MTC2 CCAG
MTC3 TTGA
MTC4 GGTCA
MTC5 AACAT
MTC6 CCACG
MTC7 CTTGTA
MTC8 TCGTAT
MTC9 GGACGT
MTC10 AACAGAT
MTC11 CTTGTTA
MTC12 TCGTAAT
They are used in place of the common adapter in the standard protocol (1ul/sample). One possible use, and simplest to use as an example, would be to use these to run 12 plates in a lane. In this case you would make a master mix for the ligation of each plate which contains a different MTC adapter.
Where are they? In the -20 at the back left corner of the bay on the bottom shelf in a box that has a pink lab tape label that says something to the extent of “barcodes + barcoded adapters 1-12”. This contains the working concentration for each of the MTC adapters. Beside that is a box containing the unannealed and as ordered oligos and the annealed stock. The information regarding what I did and what is in the box is written there. The stock needs an additional 1/20 dilution to get to the working concentration
How it looks
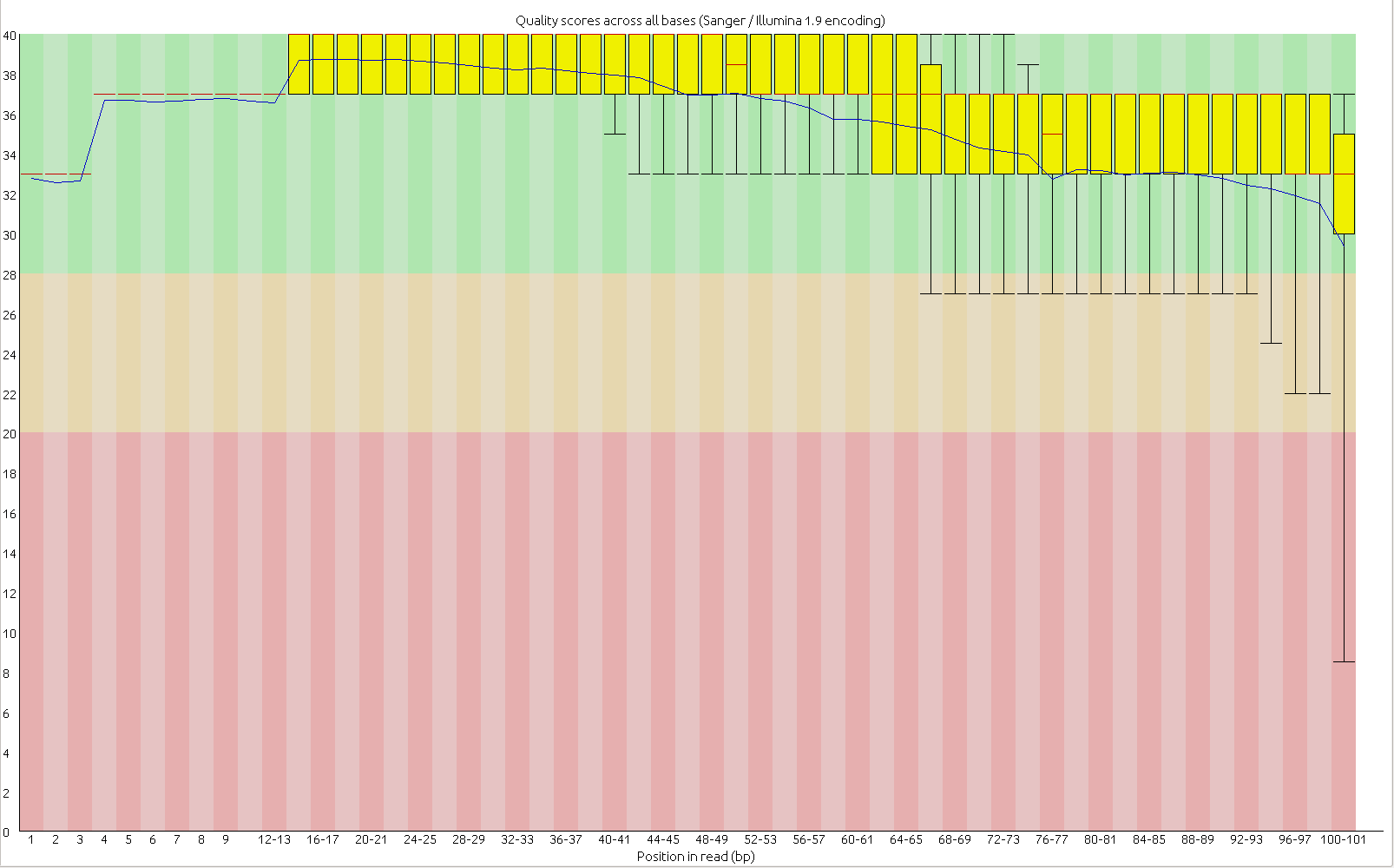
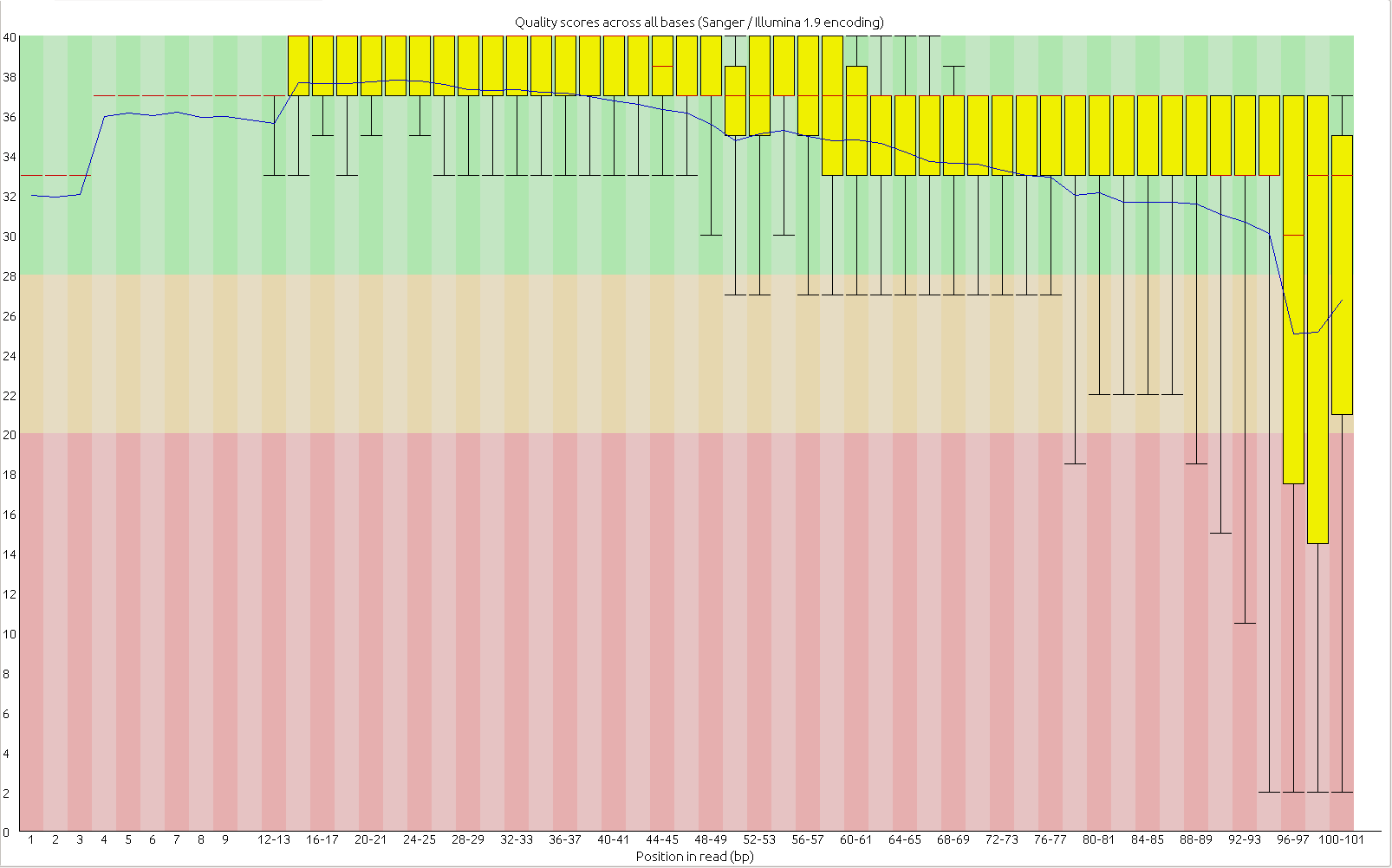
First, the quality of the second read is just about as nice as the first read. Using fastqc to look at 4million reads of some random run:
Read one:
Read two:
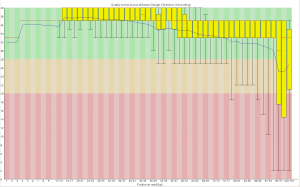
Now, for the slightly more idiosyncratic part: read counts. In short I dont see any obvious issue with any of these barcodes. I did 5 sets of 5 plates/lane. For all the plates I used the 97-192 bacodes for the Pst side. Then each plate got a differnt MTC barcode for the MSP side. Following the PCR I pooled all of the samples from the plate and quantified. Each plate had a different number of samples which I took into account during the pooling step. Here is the read counts from a randomly selected 4 million reads corrected to number of samples in that plate. Like I said it is a little idiosyncratic but the take home is that they are about as even as you might expect given usual in accuracies in the lab, my hands, and the fact that this is a relatively small sample.
Lane 1
MTC5 14464
MTC1 13518
MTC7 14463
MTC9 13448
MTC3 14232
Lane 2
MTC10 30395
MTC6 11267
MTC2 8263
MTC4 19295
MTC8 14766
Lane 3
MTC5 16631
MTC7 17315
MTC11 11623
MTC9 16256
MTC3 13831
Lane 4
MTC10 11302
MTC6 12120
MTC4 10326
MTC12 18959
MTC8 12832
Lane 5
MTC1 13151
MTC6 13490
MTC2 12851
MTC11 12460
MTC12 17296