The following protocol is for producing electro-competent cells of agro-bacterium. Currently A. tumefaciens agropine-type strain EHA-105 (Hood et al.,1995) is used for transformation of sunflower and arabidopsis. Harvesting new competent cells takes 4 days to complete with the 4th day being the most labour intensive. It’s advised to begin early in the morning on the 4th day as the length of culture, purifying, and aliquoting can take several hours.
BRC wifi on the 3rd floor
Many people on the 3rd floor of BRC building told me that their wifi (ubcsecure) keeps getting disconnected. I had the same problem, so I have asked UBC IT services for help.
IT services suggested me to do the following steps:
1)remove ubcsecure network from the wifi option
2)connect to ubcvisitor and go to autoconnect.it.ubc.ca to run the tool
3)ubcsecure is created automatically
4)reconnect to ubcsecure
Now it is working for me.
I hope it would work for everyone else in the building.
BigDye 3.1
While cleaning up the freezer a few months back we found encrusted in a block of ice at the bottom of the common lab freezer a box with a seizable amount of BigDye 3.1, the reagent used to prepare samples for Sanger sequencing (basically a PCR mastermix with labelled nucleotides). As that is expensive stuff (what we have is worth 3-4000 dollars), I tested it to see if would still work. All the tests I did were rated “great sequence” when I got the results back from NAPS, so the BigDye is fine.
Thanks to a donation of buffer from NAPS, I have diluted some of the BigDye to the same working concentration NAPS uses (1 part of BigDye, 1.5 parts of buffer, 0.5 parts of water). Follow the instructions on the NAPS website (http://naps.msl.ubc.ca/dna-sequencing/dna-sequencing-services/user-prepared/) to prepare your sample, and use 3 µl of the diluted BigDye. There are eight aliquots of about 150 µl each (50 reactions) in a box with a yellow “BigDye” label in the common -20 ºC freezer. If you plan to do only a few reactions at a time, consider making smaller aliquots for your personal use (BigDye doesn’t like repeated freeze-thaw cycles). To avoid confusion I kept the concentrated BigDye and dilution buffer in their Applied Biosystem box on the door of the first freezer on the right in the freezer room. If we run out of dilution just let me know, and I’ll be happy to prepare more.
How to do effective SPRI beads cleaning
Reply
SPRI beads cleaning is one of the most repetitively used step during library preparations and probably the step where most of us lose a lot of precious DNAs.
Losing DNA scared me so much (because it can be observable) that I hesitated a lot before trying to use beads to concentrate genomic DNA, because usual rate of recovery are ~50%.

Hopefully with some practice and a lot of patience, it is possible to reach 90% recovery. How to lose as little DNA as possible? Here are some guidelines: Continue reading
SB vs TAE: Fight!
Since I started in the lab, I had the impression that the SB buffer we use for gel electrophoresis did not work quite as well as the more commonly used TAE buffer, but as I wasn’t running many gels I was too lazy to run a test. Also, Wikipedia said I was wrong, and who am I to doubt Wikipedia?
Clean and cheap DNA from argophyllus (and other sunflowers)
Argophyllus has a reputation of being a plant it is hard to get DNA from. As a test for a larger project, I did a round of extractions from annuus, petiolaris and argophyllus. I used the modified 3% CTAB method I described before, starting from one fresh, frozen, very young (~1-2 cm long) leaf.
For all species, the CTAB extraction yielded about 50 µl of 200-500 ng/µl solution (10-25 µg in total) of clean (260/230 = 2.05-2.20) genomic DNA, with minimal shearing (see picture). Continue reading
Bike trailer available
This is just to let everybody know that for those that are working around campus this summer, we have a bike trailer that can be helpful to transport small equipment or a bunch of plants from the lab to the greenhouse, Totem field or anywhere near by. There is also a lock for it and hopefully, and most importantly we will soon have an additional bike apart from the rusty one that Brook kindly donated some time ago. I will keep you updated on that!
Share and enjoy! (make sure to always return it to the lab so it’s available for the rest of us)
Thank you,
Natalia

GBS dual-barcode deplexer
There is a version of the GBS barcode protocol that has barcodes on both adapters. Although scripts existed for demultiplexing dual enzyme GBS (including PyRad and Stacks), it didn’t seem like any of them let you demultiplex for dual barcodes. For this you need you need to determine the sample identity by both barcodes (i.e. either barcode may not be unique) and you need to strip out barcode sequence.
Drill, dry, sieve
I wanted to let everyone know about some supplies and equipment that are new in the lab. During the 2015 field season, I bought a couple of things that are now part of the lab general use equipment and supplies. Here they are:
- Drill. We now have a nice, battery-powered drill that is really good for drilling things. The drill is in the hardware cupboard near the lab computer. It’s in an orange bag.
- Silica Gel. I returned from the field with more than 50 kg of used silica gel. It’s not pristine (was used to dry seeds), but great for field work. It’s stored in sealed, orange buckets in the Lab Extension in Biology (check the tall metal shelves).
- Seed Sieves. For processing seeds in Iowa, I bought some seed sieves. These are supposed to be the best ones for wild Helianthus, so if you are processing a lot of seeds, these might be of use. These are also now in the Lab Extension.
Happy drilling, drying, and sieving.
Phytochemical databases
If you are interested by generating quickly a list of compounds that were previously identified in a plant, the online dictionary of natural product is a good place where to start your research. It regroups entries for >270k natural chemical compounds.
In the current version of the website (March 2016) you can search for “Biological source” and in there “the latin name of your favourite species”. The result of such a query for “Helianthus” is a list of 381 chemical compounds that were tagged as identified in Sunflower species (and a minority of other species with helianthus in the name). Of course this list is quite restricted compared to the number of metabolites produced by sunflower plants. It is however a good starting point to know what kind of compounds (here a lot of terpenes, few flavonoids) were previously extracted from your favourite plant species. Some entries are tissue specific, such as large terpenes from pollen. Also interesting, the “biological use” column gives sometimes information on the role of the compound (e.g. antifungal, allelochemical, growth inhibition).
The CAS registry number is probably the most powerful piece of information you can get out of such a search. It’s the unique ID for the compound and can be use to search the compound in multiple databases. My favourite are:
- Chemspider: This is probably the more powerful chemical database that currently exists. It’s huge: it regroups information for more than 44 million chemical structure and cross-reference with many other databases.
- Pubchem: It will look really familiar to NCBI/pubmed users. With pubchem you can for exemple submit the CAS number and get a list of publications linked to the compound.
How to do GBS libraries with “difficult” DNA samples
First of all, let’s be clear about it: Having good amount of high quality DNA should be a starting point for all projects. Recently, we had this conversation at lab meeting about the “one rule” to succeed in establishing a lab (quoting Loren): “Don’t try to save money on DNA extraction. Working with high quality DNA reduces cost at all downstream steps, even on bioinformatics”.
However, if you need to work with “historical” DNA samples from the lab (I genotyped old DNA plates at least 8 years old) or DNA from collaborator for which you have no control over the DNA quality and/or no more plant tissue to redo DNA extraction, here are some tips on how to get a maximum out of (almost) nothing.
I started the GBS protocol with 100ng of DNA, it works. However, if you want to save yourself a lot of time and the lab some money on repeating PCR, repeating samples, repeating a lot of qubit measurement, start with 200ng.
A) If some of your DNA samples are <8.5 ng/ul (100ng protocol):
Among the 1500 DNA samples I received from a collaborator, 134 did not meet the requirement (>8.5 ng/ul) to start the GBS. I thought about concentrating these DNA with different methods: 1) using beads: you need to be ready to loose 50% of the DNA; 2) speedvac: I did not find one (supposedly there is one in the Adams lab?) and I was concerned about over-concentrating TE in the same time as DNA.
Hopefully, if you look at the digestion step, a large volume of the digestion mix is water/tris. By removing this water, I was able to include in the protocol DNA with concentration >5.8ng/ul, recovering half of my problematic samples. Just be extra-careful when pipetting the 2.8 ul of “water-free” digestion master mix. I had good PCR amplification for these samples.
B) If you are desperate:
I used whole genome amplification (WGA) prior to starting the GBS protocol to increase the DNA concentration of “historical” DNA samples. You will probably recover most of your DNA samples if they are more than 1ng/ul.
However, DO NOT MIX genome amplified and plain genomic DNA on the same plate for sequencing, especially if you pool your library before doing PCR and qubiting. The WGA samples amplify much better and Sariel showed me libraries in which few WGA samples took a large part of the sequencing reads. It’s a recipe for disaster and high missing data.
My strategy was to qubit all the DNA plates and estimate the remaining volume. If the remaining DNA sample was less than 100ng, I did WGA but I moved these samples to specific WGA plates. It’s a bit more work because if your samples are already in plates, you will need to relocate all your samples. From my experience, it’s worth it.
Whole Genome Amplification
I used Whole Genome Amplification (WGA) to recover some very old DNA and include a maximum of samples into GBS plates for a mapping project. I will do another post about that, but here I would like to resume how I did the whole genome amplification.
For information on how the whole genome amplification method works, check the previous post by Moira.
I used the Qiagen Repli-g Mini Kits with the amounts divided by half.
The protocol is the following:
2.5 ul of DNA
2.5ul of buffer D1 (DLB + nuclease-free H2O)
Incubate for 3min at room temperature
Add 5ul of buffer N1 (stop solution + nuclease-free H2O)
Vortex and centrifuge
Add 15ul of master mix (repli-f reaction buffer + DNA polymerase)
Incubate at 30oC for up to 16h and inactivate enzymes at 65oC
I found that 6h was good for the amount of DNA I was hoping to recover. I recovered at least 50x the amount I started with, typically starting with concentrations between 1-10ng/ul and getting on average 60ng/ul out of the WGA protocol.
Depending on the use of the DNA, you may want to play with the incubation time (e.g. keep it short), especially if you plan to use PCR based approach later on (almost everything is PCR-based..).
Comparing aligners
When analyzing genomic data, we first need to align to the genome. There are a lot of possible choices in this, including BWA (medium choice), stampy (very accurate) and bowtie2 (very fast). Recently a new aligner came out, NextGenMap. It claims to be both faster and deal with divergent read data better than other methods. Continue reading
Resources for drought genes in sunflower
Recently, I hassled people until they gave me places to look for lists of genes expressed in response to drought treatment in Helianthus. Perhaps you will find it useful too?
Marchand et al. 2014 drought gene regulatory network: http://onlinelibrary.wiley.com/doi/10.1111/nph.12818/full.
Based on experimental data from Rengel et al. 2012: http://dx.plos.org/10.1371/journal.pone.0045249
And more in Marchand’s thesis: http://thesesups.ups-tlse.fr/2597/
Additionally, Min has some data/preliminary analysis from a microarray study in sunflower. Contact him for more info?
GBS barcodes 97-192
If you are using GBS barcodes 97 – 192, here is the sequence info (from Brook). This might be somewhere else on the blog, but I couldn’t find it! Also included, sequence info for barcodes 1 – 96.
New germplasm from Dylan’s 2015 field work
As many of you know, I was tasked with collecting wild populations of Helianthus annuus, H. petiolaris, and H. argophyllus from across the geographic range of the species, for use in the abiotic stress adaptation project. Over a three month period, I drove more than 30,000 miles and collected at 145 Helianthus localities in 15 western states. I found a lot of really interesting plants, including what might be several new species. In future posts, I’ll talk some more about the trip, and what I found.
The seeds are now here. They are cleaned and packaged in individual envelopes numbered by population and mother plant. I cleaned the seeds in Ames, Iowa at the UDSA facility, so they are high quality, ready to go. All of the seeds are now part of the seed collection in our lab in the Biodiversity building. The new seeds have been added to the permanent database of seeds for the whole lab. But I also put a copy of my complete notes here. My notes give details about where the plants were collected, their ecology, etc. I’ll be putting printed data in the boxes with the seeds as well, just in case the zombie apocalypse arrives and these electronic data go dark. I also plan to post photos of each population I visited, but that’s down the line. For now, the seeds are here, so if you need to access any of them for a project, the data linked to this post should help you do that. If you happen to use any of these plants in a project, please note that voucher specimens were deposited in the herbarium at Indiana University (herbarium code: IU).
If you need to contact me about these seeds, and you have no idea who I am or where I am located, here is my “permanent” email address, which should also last for some time.




Using Monsanto Acceleron DC-309 fungicide for seed treatment in preparation for export
Greg Baute procured a small bottle of, what I believe to be, Monsanto-manufactured Acceleron DC-309 for fungicidal treatment of seeds in preparation for export. It wasn’t labeled as Accelron, but did have a seed treatment label for a related chemical taped to the outside. Based on Greg’s earlier emails and with this information, and Winnie’s confidence, I wrote “Acceleron” on the bottle in Sharpie. It’s a small red bottle. Should you need to use it, Winnie typed up the following protocol. There are links to safety documents at the end of this post. Be sure to wear Nitrile, not natural rubber, gloves.
How to apply fungicide to Greg Baute’s sunflower seeds
- Make sure you wear gloves!
- Take a falcon tube, a dropper, the fungicide and sunflower seeds to the fume hood
- Put sunflower seeds into the falcon tube (depending on the size of the seed, you might need to grab a 25mL/small one for small seeds or 50mL/large one for larger seeds)
- Use a dropper and put 3-4 drops of fungicide into the falcon tube and seal the lid
- It is now safe to bring the falcon tube with the fungicide and seeds out from the fume hood. Use the vortex to mix the fungicide and seed. Shake them for about 10-15 seconds.
- Return to the fume hood and open the lid, take seed out.
- Repeat for next package of seed
Have fun! But please be safe.
Chemical’s home page: http://www.acceleronsts.com/Corn/Pages/Resources.aspx
Seed treatment product label: http://www.acceleronsts.com/Corn/Documents/DC309.pdf
EPA Chemical Resistance Category Selection Chart: http://oeh.cals.cornell.edu/sites/oeh.cals.cornell.edu/files/shared/documents/pesticides/EPAChemRes.pdf
H. petiolaris collecting trip – GSD 2015
Kate and I travelled to Great Sand Dunes (GSD) National Park in Colorado for a Helianthus petiolaris collecting trip this September (14th to 19th). We collected seed (and in most cases, leaf tissue) from ten established sites and five new populations. Continue reading
hand over documents
I’ve put together a folder containing information for anyone who may want to continue any of the projects I worked on. I’ve zipped it into a archive called “HandoverDocument_GBaute.zip” and it can be found on /moonriseNFS/ on a external harddrive that I am going to give to Loren and on a couple of thumb drives I am leaving in the lab (one is with my lab notebooks another is in the drawer by Winnies/Megans desk area). You will find information on the seeds, DNA and data used in my thesis and in a number of other projects.
hand emasculation of sunflowers
So you want to do some crosses without CMS or relying on self incompatibility? It takes some dedication but it can be done! This is how I carry out hand emasculations. There are hormone based options out there but they can need background specific protocols.
- Grow your plants.
- Bag the heads you want to use when there is 2-3cm between the last leaf and the head. Don’t bag too early and don’t bag too many leaves this will make the head grow crooked.
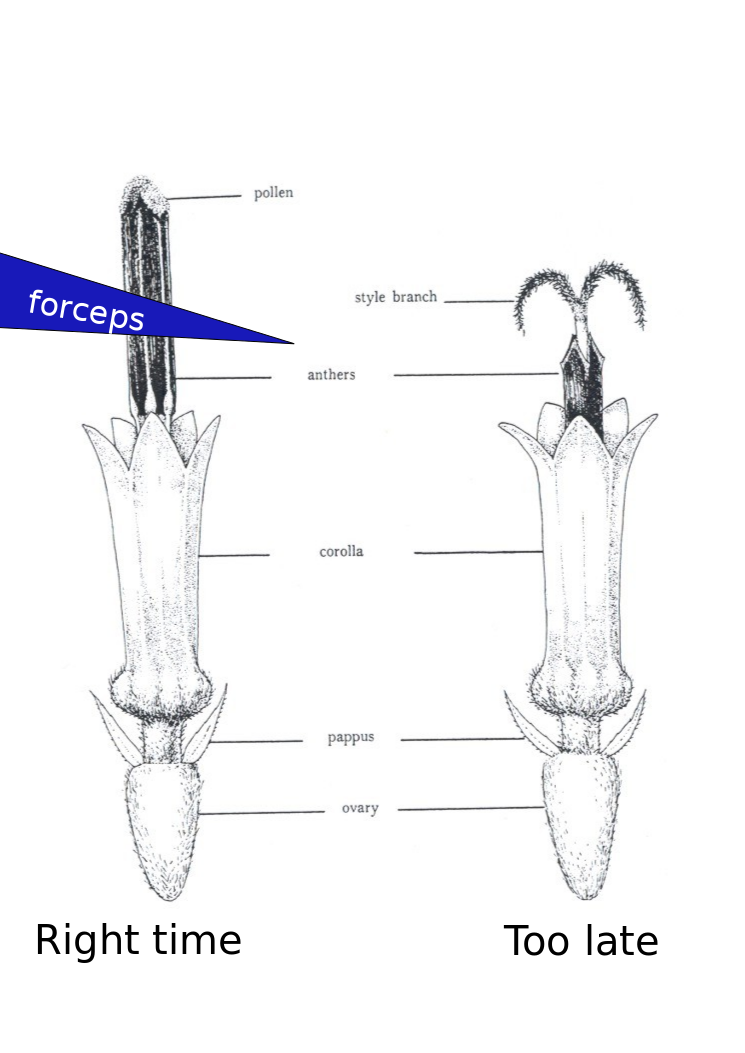
- Show up ~30 minutes after sunrise or when the greenhouse lights come on. There is variation here with the temperature effecting when the anthers come out of the florets. You can usually figure out the right time to come in a few days. If you show up to early you will only be able to see the very tips of the anthers as the come out of the corolla. These are very difficult to remove. Waiting 15-30 minutes will make a big difference. You will also notices some lines progress at different rates than others.
- Use a set of tweezers like these: http://www.aventools.com/avens-complete-product-line/tools/tweezers-and-quick-test-tweezers/precision-tweezers-1/e-z-pik-precision-tweezers/e-z-pik-tweezers-7-orange#.UzxaJ63c-d8 to remove that days new anthers. See figure for about where and when you should grab the anthers. You can grab a few at a time. You will notice as the morning progresses the pollen at the top of the anther will start to come out and fall all over the place. I find there is about a 1 hour window which is optimal.
- Use a good spray bottle set to a ‘jet’ spray (not mist) and spray down the head. This step is very essential.
- With the head wet inspect it for anther fragments either still in the corolla or fallen down in between florets. Repeat steps 4+5 as needed.
- Repeat steps 3-6 until the flower is done. I find this is about 1 week to ten days for a large cultivar head. If you don’t want to wait for the every floret you can use the forceps or a knife to remove the center florets.