This post is about fragmenting, or shearing, genomic DNA to a particular size range using the Rieseberg lab’s Bioruptor sonicator.
Most of the current whole genome shotgun (WGS) library preparation protocols for NGS applications start with fragmented DNA. Generally speaking, this starting DNA should be a certain size and, for multiple samples, consistently that size. This objective turns out to be quite a tricky thing to accomplish with the Bioruptor. Given that WGS sequencing will probably continue to be popular in the lab, I am posting here what I have learned so far about taking whole genomic sunflower DNA and smashing it to the size range that I want using the Bioruptor. If I discover anything else in future library preps I’ll add it below. If anybody else has useful tips please comment.
What you want for a WGS sequencing library
- DNA fragments of size suitable for your sequencing strategy. For example, WGS libraries for Illumina paired-end sequencing (on the Biodiv. HiSeq for instance) generally start with fragments around 300-400bp, (which become longer with the addition of the Illumina adapters).
- A representative sample of the genome. This is critical – usually a WGS sequencing library is assumed to represent the genome. That is, the fragments that get sequenced are assumed to be a random sample of the genome. (Note that there might be strategic reasons for wanting a biased WGS-like library in some cases, see below.)
So, obviously, the very first step in a WGS sequencing library preparation, DNA fragmentation, is critical. To reiterate, you want fragments of the right size and you (usually) want them to be an unbiased sample of the genome. See my comments at the bottom of this post for more about bias.
Sonication, the Bioruptor and Alternatives
Sonication breaks DNA molecules and is what the Bioruptor does. Sonication is a relatively unbiased way to shear DNA, and with the Bioruptor its cheap too. Note that there are other ways to shear DNA. There are enzymatic methods and nebulization, and then there is the Covaris. As far as I understand it, at this point, enzymes and nubilization are not highly regarded for WGS sequencing libraries while the Covaris method is the gold standard. Search SeqAnswers for “Covaris” to find out more.
Covaris: The Covaris might be thought of as technologically tricked out sonication. It is inherently better than sonication with the Bioruptor but the machine is much more expensive (on the order of $60K CAD versus something like $15K CAD for the Bioruptor) and the per-sample consumable cost is also much higher (on the order of $5 CAD versus a few cents for the Bioruptor).
NAPS has a Covaris machine. NAPS, however, seems to be run with a fairly hostile view of the client and, as of November 2011, they are asking $10 CAD to shear a DNA sample with their Covaris machine and they are not prepared to allow hands-on access to the machine such that we could provide the labour and reduce the cost. The NAPS Covaris machine is not a realistic option for many WGS sequencing projects in our lab due to the cost-benefit equation but if you are preparing just one or a few samples it might be economically viable. It might be worth paying NAPS for Covaris shearing if you are preparing only a few libraries and accurate fragment size and low sequence bias are highly important to you, especially if you are in a hurry.
Bioruptor

We have a Diagenode Bioruptor sonicator in the lab. Its in the support room off our lab on the 3rd floor of Biodiversity where our fridge is. Our model is the BioruptorXL.
You can download a pdf of the manual for it here.
And you can see Diagenode’s webpage on it here.
And you can download a not overly useful one page DNA shearing protocol from Diagenode here.
The Bioruptor is a pretty simple machine. Its a programmable sonicator with a standard water bath but also with a simple tube circulating apparatus consisting of a small electric motor that rotates the two tube racks via plastic cogs, (see the blue circular things through the window in the image above).
The programmable aspect of the Bioruptor allows the operator to set the sonication intensity, the total time of sonication and the on/off interval for a period of sonication. Note that it seems to be normal to sonicate DNA with an on/off timing such that the sonication is on for a period then off for a period repeatedly over the total sonication time.
Sonication intensity: There is a dial on the control box. You can set it to Low (L), Medium 1 (M1), Medium 2 (M2) and High (H). For DNA shearing Diagenode recommends Low power but I have been doing my DNA on High (see below).
Times: There are three time parameters that are set via the little LCD screen and associated buttons on the control box. Navigate the menu using the arrow buttons and the “Ok” and “Esc” buttons (see the manual if you get stuck). T1 is the total sonication session time. You might set this parameter to 2 minutes or 5 minutes or 20 minutes for example. T2 is the sonication “on time” and T3 is the sonication “off time”. You might set both of these to 30sec for example.
If you set T1 to 10min, T2 to 30sec and T3 to 30sec then pressed the Start button, the Bioruptor would run for 10 minutes sonicating for 30 seconds in every minute for a total of 5min of sonication over the 10 minute run.
Cooling: Sonication generates heat. Heat is bad as it can exacerbate sequence biased shearing (see below). So, you need to cool the water bath while you sonicate DNA. The Bioruptor manual recommends using a water refrigerator and a pump to circulate cold water through the bath, or floating a layer of ice in the water bath. We don’t have the refrigeration accessory. I have, so far, not done the ice layer thing (see below). Instead, I fill the water bath with ice at least 10mins before I want to start and I keep water in a big flask in the fridge. I put a few generous handfuls of ice in this water, mix it up then scoop out the ice that is pre-cooling the bath and fill it with the iced water. I then scoop almost all of the remaining ice out of the water bath before I start sonicating. So, contrary to the manual’s recommendation, I do not have a layer of floating ice in the water bath but the water bath and the water start out ice cold. I change the water every 10 minutes as well. I explain why I don’t use a layer of ice below.
Number of Samples/Tubes: We have a pair of tube racks each of which takes 12 1.6ml tubes and another pair each of which takes 24 0.65ml tubes. These pairs of racks can be used together so the maximum number of samples that can be sonicated together is 24 if you use 1.6ml tubes and 48 if you use the small tubes. The number of samples in the machine at once is one of the variables that affects shearing, (see below).
Predictable shearing: Unfortunately shearing DNA with the Bioruptor is not as predictable as one would like. There are a number of variables that influence how effectively DNA is sheared, the sonication power and time, obviously, but also: the number of samples in the machine, the volume of each sample, the DNA concentration of each sample, the type of tubes and the temperature.
What I do
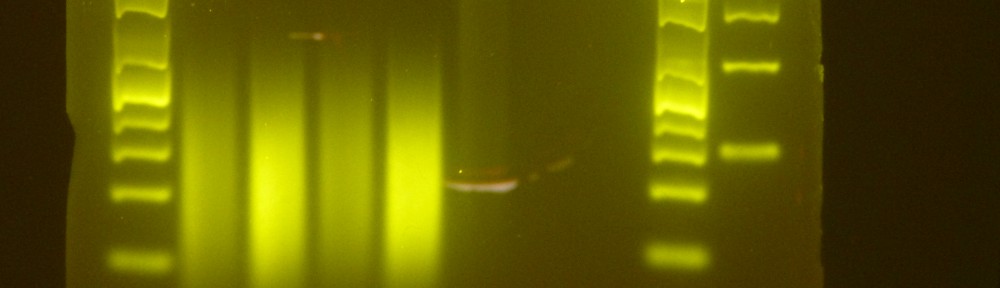
As of January 2012 my strategy for shearing DNA for Illumina WGS sequencing libraries is to process 12 tubes at a time; running them in the right hand side tube rack with nothing in the left rack; using Costar 0.65ml tubes with 3ug of genomic DNA in 75ul of water; sonicating for 15mins with 30secs on and 30secs off then 10mins with the same on/off times with an iced water change in between. I then run out 2ul of each sample against a ladder on 2% agarose to check the shearing. Any samples that are insufficiently sheared go back for another 5mins and then 2ul is run out again and this is repeated as necessary. I have experienced a low frequency of samples being overly sheared after the initial 15min+10min sonication and when I have repeated these samples with a 10min+10min sonication they have been better.
Note that I always have 12 tubes with 75ul of volume in them. So, if after the first 25mins of sonication I have fewer than 12 tubes that need further sonication, I make the batch up to 12 with dummy tubes with 75ul of water in them.
See below for some examples and images.
Some experiments and observations
- The rotating tube racks can get stuck! This is bad. The location of the tubes in the waterbath seems to dramatically affect shearing, such that a tube in one position can be much more thoroughly fragmented than a tube elsewhere. Presumably this is why the Bioruptor has rotating tube racks. After some mysterious, and substantial, within batch variation in shearing I stood and watched the machine while it was running and saw the tube racks get stuck and stop rotating. I fiddled with the racks and watched them rotate and get stuck a number of times but I’ve never figured out what the cause is or how to prevent it. My most recent experience (shearing 42 samples so far in Oct-Dec 2011) has been good – no getting stuck but I still keep an eye on the rotation. If you get wild variation in shearing I’d suspect the rotation mechanism. If your DNA is particularly precious, i.e. you can’t afford to ruin it and start again, I’d stand and watch the Bioruptor in operation. You can open the door and force the racks to rotate by hand although the noise is a little annoying and possibly bad for you – I’d wear ear plugs if you have to do this.
- Some samples atomize! I haven’t worked out why this happens although it does seem to be repeatable in that the same sample will atomize to some extent in multiple bouts of sonication. The atomization, particularly when its bad – i.e. there is virtually no volume at the bottom of the tube, its all in tiny droplets – seems to dramatically decrease the shearing effect of sonication. If anybody figures out why this happens or what to do about it please let me know and comment on this post. Whenever I sonicate samples in multiple bouts I spin the tubes down between bouts.
- The number of tubes in the machine matters. I spent some time optimising sonication time for four or five samples – aiming for a smear centered at 400bp, I landed on 13mins on High with 30sec on/off cycles and a layer of floating ice in the water bath. I then tried my optimized strategy with 12 samples and found that I had to sonicate them much longer than 13mins. Take a look at this image and this image for examples of samples sheared in batches of 4-5 for 13ish minutes and compare with this image of 12 samples sheared as a batch for 13 minutes. Unlike the 4-5 sample batches, the 12 sample batch is not even close to adequately sheared. Note that the ice may be part of the problem, see below.
- DNA concentration doesn’t appear to matter, at least within a certain range. Take a look at this image of two samples sonicated at two different concentrations in the same volume – 2ug or 5ug in 50ul, i.e. 50ul volume of 40ng/ul and 100ng/ul DNA. Note that there were only 4 tubes in the Bioruptor for this test (perhaps concentration might matter with more tubes??) and that I took 2ul out of the tubes at 5 time points – so at the fifth time point there was only 42ul (who knows if this matters).
- Ice vs no ice. As mentioned above I optimised sonication in the bioruptor for 4-5 tubes with a layer of floating ice as per the manufacturer’s recommendation. Take a look at this image of a test of sonicating with and without floating ice in the waterbath. The ice seemed to have no negative effect on fragmentation. However, when I stepped up to 12 tubes in the Bioruptor at once the ice appeared to be part of the cause of the poor fragmentation. Compare this image of 12 samples sonicated for 13mins with ice with this image of the same 12 samples sonicated for 13mins without ice.
- 12 sunflower genomic DNA samples (11 annuus, 1 maximiliani) sheared for WGS paired-end sequencing library prep for Illumina HiSeq in October 2011 by me (Dan E.). These are the first samples I sheared and made libraries for. Take a look at this image to see the final sheared DNA samples that went on to the first step of library prep. Note that it was a poor gel – electrophoresis was distorted by a piece of plastic I used to hold the gel straight. Ten of these samples have a DNA smear centred at 400bp which was my objective. Two of them have a DNA smear centred somewhere between 300 and 350bp, which is sub optimal. Three micrograms of these 12 samples in 75ul were sonicated on High with 30sec on/off cycles with no ice for 10min+10min+10min, 15min+10min or 15min+10min+5min with iced water changes between bouts. I’ve annotated the gel image linked above with the sonication times.
- 30 H. petiolaris genomic DNA samples sheared for WGS paired-end sequencing library prep for Illumina HiSeq in December 2011 by me (Dan E.). These were the second batch of samples I prepared for sequencing. Based on my experience with the first batch of 12 samples I processed these samples in batches of 12 sonicating on High with 30sec on/off cycles with no ice in the waterbath but with a pre-cooled waterbath, ice-cold water and water changes between sonication bouts. I started with 3ug of DNA made up to 75ul with water in 0.65ml Costar tubes (same as above). This is how it went:
- I sonicated all 30 samples (in sets of 12 with dummy’s to make up numbers) for 15min+10min then ran 2ul of them out on a 2% agarose gel – see this image and this image. At this point I set six of the 30 samples aside. Five of these were nicely fragmented, i.e. the DNA smear was centred at 400bp, while one was a little over done, i.e. the DNA smear was centred at <400bp.
- The remaining 24 samples were sonicated for an additional 5mins and then 2ul was run out on 2% agarose – see this image and this image. After 15min+10min+5min sonication 16 of the 24 samples were nicely fragmented (so, 21 of 30 finished at this point) but 7 of the 24 were slightly over-sheared (so, 8 of 30 are now a little too fragmented), and one sample remained insufficiently fragmented, i.e. centre of DNA smear was >400bp.
- Of the 7 overly sheared samples I had sufficient DNA to repeat four. These four samples, again 3ug in 75ul, were sonicated for 10min+10min. The single sample from the original sonication bouts that remained insufficiently fragmented was given an additional 5mins (so, 15min+10min+5min+5min). Two ul of these five samples was run out on 2% agarose – see this image.
- So, after several rounds of sonication in the Bioruptor and a few second attempts I had 30 sheared DNA samples ready for library prep. Note that it took some days and the 30 sheared samples were not perfect – there remained 3 samples that were slightly over-sheared and there were 2 that were probably under-sheared. See this image and this image for the final 30 sheared samples that I moved on to library prep. Shearing with the Bioruptor is not controllable enough to expect perfection over numbers of samples. To my mind the objective is to get as consistent results as possible. {Remember that if this inherent variation in fragmentation is particularly problematic for your purpose you might want to pay the money for Covaris shearing.}
What you should do
- Work out a strategy that balances the time it takes to get what you want with the risk of overdoing it and having to start again (assuming you even have that option). One strategy, based on my experience so far, would be to sonicate in batches of 12 in 0.65ml Costar tubes, in the right-side rack, on high with 30sec on/off cycles and no ice in the waterbath for 10min+10min, then check the shearing on agarose. Then do additional 5min bouts for insufficiently sheared samples – checking on agarose after each bout and removing adequately sheared samples as you go (replacing them with dummy tubes containing water so there are always 12 tubes in the rack).
- If you want to do some experiments with the number of tubes or the sonication intensity or with floating ice in the waterbath, please go right ahead and tell us how you go in the comments on this post.
- Keep an eye on the rotation while the Bioruptor is running.
- Check your samples for atomization and spin them down between sonication bouts.
A word on bias in WGS sequencing libraries
A WGS sequencing library is a sample of DNA fragments from a genome. That sample is biased if the sequences of those fragments do not accurately reflect the genomic sequence, i.e. sequences from the genome are over or under represented in the fragments that comprise the library. There are two major sources of such bias – fragmentation and PCR, (currently most WGS sequencing library preps include a PCR step). Sequence bias can be introduced at the fragmentation step if the fragmentation, in terms of where the DNA molecules are sheared, is not random with respect to sequence. An extreme example is restriction endonuclease fragmentation which is explicitly non-random with regard to the sequence where DNA molecules are sheared. Any fragmentation method that is non-random with regard to the sequence that is sheared will bias a library to some extent. Furthermore, sequence-based shearing bias does not have to be extreme to bias a library, bear in mind that most WGS sequencing libraries are comprised of masses of DNA fragments – even subtle fragmentation biases will be realised in a library due to the simple immensity of the sample size.
The sorts of sequence-based fragmentation biases that might be important, even for nearly random fragmentation methods, are things like bias toward A/C or (less likely) G/C rich sequences or bias toward a sequence that occurs in an abundant repeat family, or commonly in low complexity sequence.
Note that a sequence-based bias in fragmentation might bias a library differently depending on the severity of the bias and the abundance of the sequence in question. Imagine a sequence that is much more likely to shear than the genomic average. If there are regions of the genome where that sequence is very frequent, those regions might be sheared so small as to be absent from the library. On the other hand, if the highly “shearable” sequence occurs at lower frequency elsewhere in the genome then fragments from those regions will be over represented in the library – the highly “shearable” sequence will effectively enrich the library for fragments that begin or end (or begin and end) with the shear-prone sequence as such fragments will outnumber those that terminate with sequence that has the average “shearability”.
Positive Bias (?)
My remarks here are largely to do with minimizing sequence-based bias in WGS sequencing libraries but its not hard to think of cases where bias would be desirable. An obvious example would be deliberate biasing a library away from repetitive and low complexity sequence in an effort to enrich a library for genic regions of a genome. At this stage, I’m not aware of library prep DNA fragmentation strategies for deliberately biasing libraries by sequence (enzymes??) but I wouldn’t be surprised if they are out there now or turn up in the future.
Dan E. January 2012.