A few weeks ago Kristin posted a nice blog entry on home made SPRI beads, which effectively replace the ridiculously expensive commercial AMPure beads. (Editor’s note: see this post at RLR for more about the AMPure beads). Dan already ordered the ingredients to make them in our lab as well, and when they all arrived I volunteered to prepare and test them. Following are some results. If you don’t feel like reading through the whole post, the bottom line is that they work very well, and that you are very welcome to use them (they are in an aluminium-wrapped Falcon tube in the fridge, with a white “SPRI beads” label).
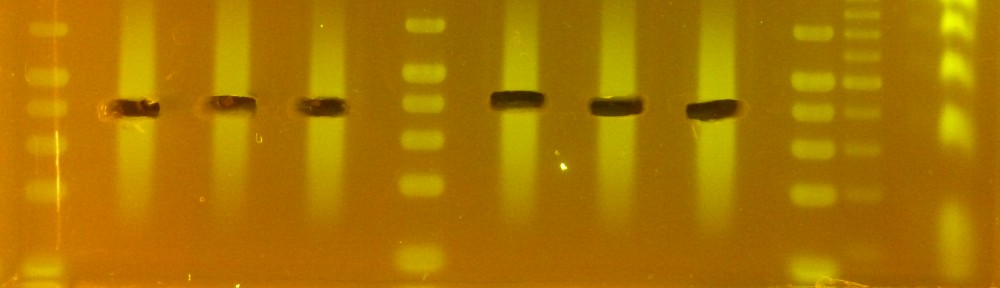
To prepare the beads I followed the protocol that Kristin posted. To test them I used as input DNA 1μg of 50 bp NEB DNA ladder in 40 μl of TE buffer, and compared them to commercial AMPure beads (Figure 1).In both cases I added to the DNA 1.2 volumes of beads. The “ladder” lanes in this and all following gels are loaded with 1 μg of 50 bp NEB DNA ladder (that is, the input DNA in most cases). All the lanes are form the very same gel, but I removed some less interesting samples in the middle.
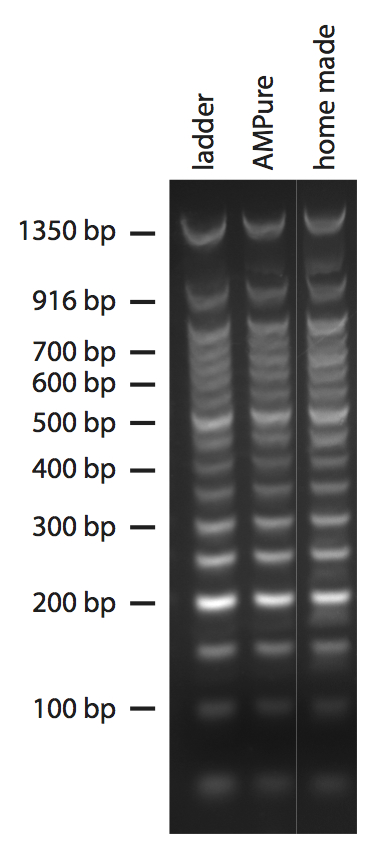
Figure 1: DNA recovery comparison between commercial AMPure beads and home made SPRI beads.
As you might know if you used SPRI beads before or if you read Kristin’s post, is it possible to use SPRI beads not only for DNA purification but also for size selection. SPRI beads are resuspended in a buffer containing PEG + salts, which facilitate the precipitation of your DNA fragments on the beads. By changing the concentration of PEG in your sample you can control the size of the fragments that will precipitate on the beads (at lower concentrations, only larger fragments will precipitate). The way in which you practically achieve this is by adding different amounts of beads suspension to your sample. In Figure 2 you can see what happens when you add various volumes of beads, from 2.1 to 0.5 times the initial volume of your sample. The upper gel shows the DNA fragments that precipitate on the beads, the lower one the fragments that remain in solution.

Figure 2: Size selection with SPRI beads, using a DynaMag magnet. The upper panel shows the DNA fragments that precipitate on the beads, the lower those that do not.
In the process of making these tests I also learned a neat trick from Ana to reduce beads carry-over, which seemed to be one of the main problems people had with our magnet. You put the tube in which you want to transfer the supernatant in the magnet as well, put the tip into it leaning on one of the sides of the tube and wait a few seconds; the beads will gather on the side of the tip and will stay there when you slowly pipet the liquid into the new tube. Well, maybe you guys already knew this trick, but I didn’t and it made me happy, so I thought I would mention it.
Next, I tried to do a double-size selection. As input DNA in this case I used both the 50 bp ladder and some H. anomalus DNA that I sheared in the Bioruptor, aiming to have a broad distribution of fragments. In both cases, I started with 1 μg in 40 μl of TE buffer. I used three different combinations of beads volumes centred around the volumes that are recommended in the Bioo protocol for size selection after shearing when aiming for 350 bp fragments (Figure 3). In the gels, the “selected” lanes show what you would keep after double size selection, and the “discarded” what you would lose in the process. I guess the concentrations of PEG and salts in the commercial AMPure beads are a bit different, because I obtained fragments centred more around 400 bp.
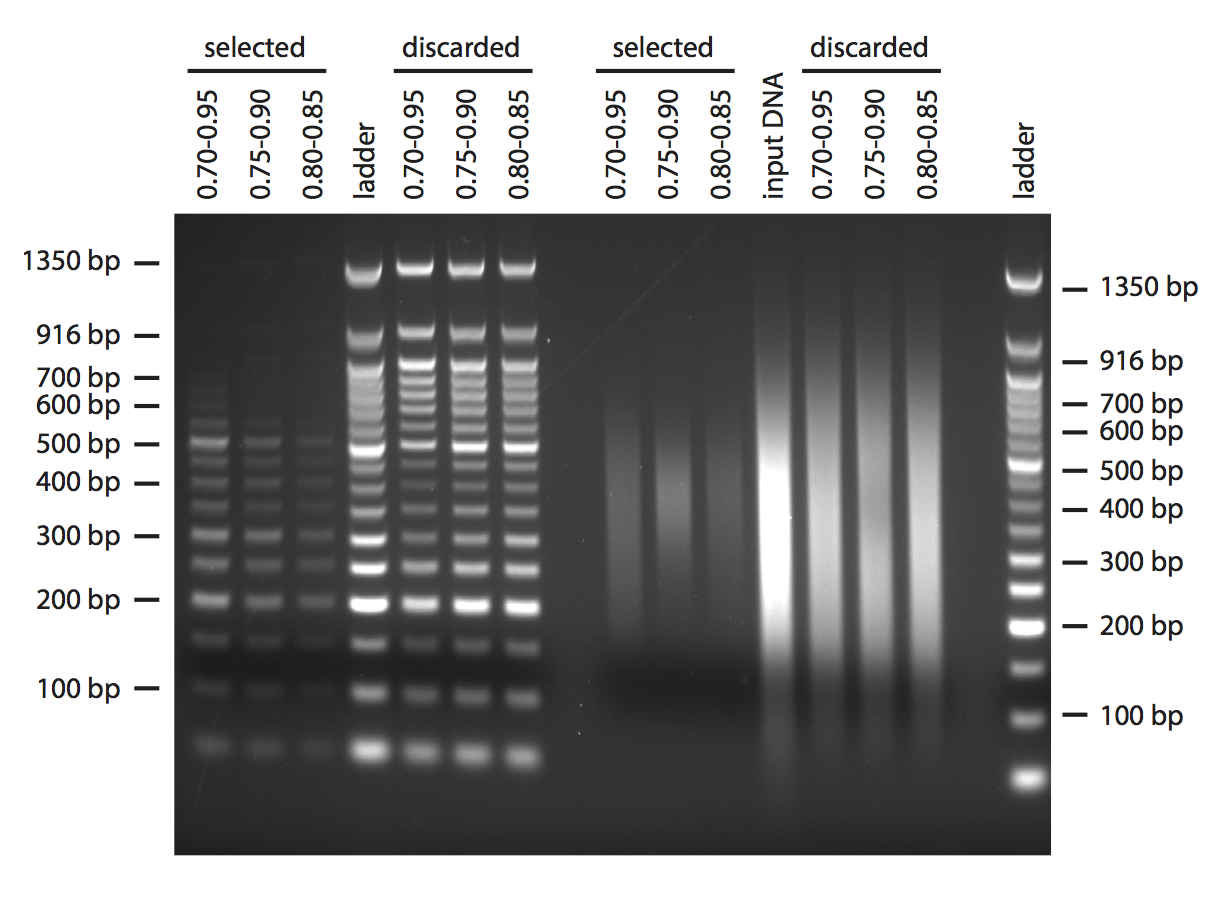
Figure 3: Double size selection.
Finally, I tried a more real-life scenario with some H. anomalus DNA I had previously sheared using the Covaris (the setting of the Covaris were such that the fragments should already be centred around 350 bp). I used two different combinations of volumes of beads, one as in the previous gel, one adjusted to aim at 350 bp fragments (Figure 4).
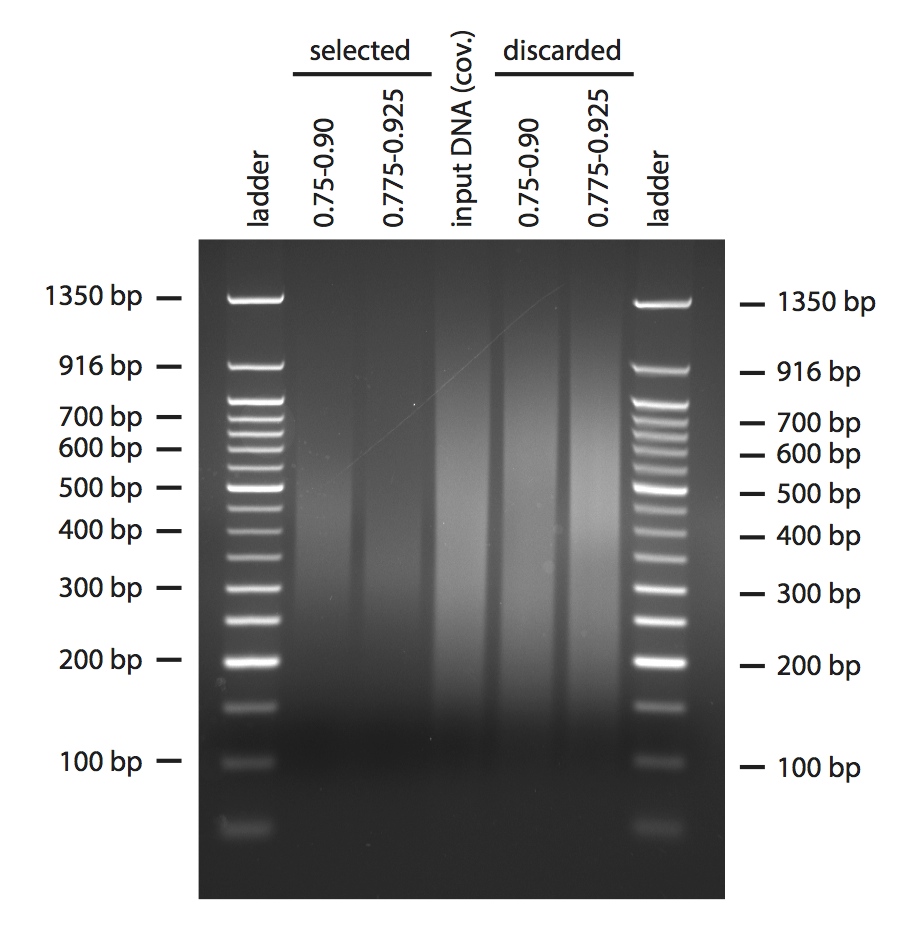
Figure 4: Double size selection on Covaris-sheared DNA.
In conclusion, double size selection works well, but you lose quite some DNA. I am not sure if it is less efficient than cutting a band from a gel, but it’s for sure faster. If you need different fragment sizes from the ones I used here, you should be able to modify the volumes of beads to use accordingly using Figure 2 as a reference. If the DNA you recover is not sufficient, you can either make a less stringent size selection, use purification from gel instead, or just start with more DNA.
As a final bonus, as I mentioned before, what makes the difference in terms of which fragments you precipitate is the concentration of PEG, not the actual amount of beads. So, if you already have beads in your tube you can precipitate again DNA of them by just adding more PEG buffer. This is exemplified in the last gel (Figure 5); there I purified 1 μg of 50 bp ladder with 0.2 volumes of beads + 1.9 volumes of PEG buffer, 0.4 volumes of beads + 1.7 volumes of PEG buffer, and 2.1 volumes of beads. As you can see, the amount of beads makes no difference in terms of size selection, but affects the amount of DNA you recover (so you need a certain amount of beads for getting a decent amount of DNA back). You can find a 18% PEG buffer in the fridge as well, in an aluminium-covered Falcon tube with a white “PEG 18%” label. In case for some reasons you need to use small volumes of beads (like the ones used in some alternative protocols for double size selection), I also made a small aliquots of 2X concentrated beads (that is, twice the amount of beads in the same PEG buffer).

Figure 5: what matters for size selection is the concentration of PEG, not of actual beads – but the amount of beads affects your recovery efficiency.
That should be it. There are about 45 ml of beads ready right now, which should last for a while. I think it would be a good idea to do some of these tests every time a new batch is prepared.
Let me know if you have some questions or if some information is missing.
Hello everybody – this is Dan E. here. See my comment below about using Marco’s SPRI beads. I tried but failed to embed an image in my comment so I’m sneaking it in here. Below is an example of an Illumina WGS sequencing library, made using Marco’s SPRI beads, run out on the Bioanalyzer.

Hello Marco and everybody else.
In June 2013 I used Marco’s SPRI beads to do cleanups and size selections for five WGS sequencing Illumina libraries. I used the 18% PEG, 1M NaCl, 0.05% Tween formulation that Marco made and didn’t tweak it with extra PEG or anything else.
They worked perfectly. (See the Bioanalyzer chromatogram I added to Marco’s post above.)
I wanted a size selection for ~350-450bp and on Marco’s recommendation I used the beads at 0.875X volume for the first step (where you keep the beads and make the lower size selection, discarding smaller DNA fragments) and at 0.825X volume for the second step (where you discard the beads and make the upper size selection). These volumes performed a very nice size selection – my libraries look good on the Bioanalyzer with nice clean peaks. The mean length for all five libraries was very close to 500bp which means that the size selection, performed on the DNA fragments before the adapters were ligated to them, was for an average length of ~380bp. That is really good.
Thanks Marco.