
The next step in the pipeline after the alignments are conducted and adjusted is to call the SNPs and genotypes. There are many programs that will do this such as the ML script that many people in the Rieseberg lab use, freeBayes, mpileup and bcftools, as well as the GATK UnifiedGenotyper. To bench mark the alignment steps in these SNP calling blog posts I used mpileup and bcftools. For example:
Category Archives: Scripts/Programs/Code
SNP calling IV – Base quality score recalibration
The best practices protocol from Broad highly recommends base quality recalibration (http://gatkforums.broadinstitute.org/discussion/44/base-quality-score-recalibration-bqsr). Apparently, the base quality scores off the machine are not very accurate and can provide false confidence in the base calls and consequently influence SNP calling. This tool will adjust average quality scores and also adjust the scores depending on the machine cycle and sequence context (i.e. start versus end of read, type of dinucleotide repeat). After recalibration, the quality score is closer to its actual probability of mismatching the reference genome.
To run this you need a database of known SNPs so the tool can assess the error rate at other sites based on the actual sequence data. Unlike humans, which this tool was designed for, most species do not have comprehensive SNP databases. However, such a table can be created by identifying SNPs in a preliminary run without the base quality score recalibration. I included any SNP that had a Qual score of 20 in my SNP vcf file.
You can see below that it had a large effect on the number of SNPs that I called. With the BAQ filter off 263,360 SNPS overlapped between the methods. 14,591 were unique to the recalibrated alignments and 131,354 were unique to the alignments without base quality recalibration.
SNP calling III – The indel problem
SNPs can often be falsely called around indels. In particular, if indels are near the start or end of a read the read is often incorrectly aligned. Here is an example of how things can go wrong from a spruce alignment. Below, at site 109 there is a TT insertion in the reads relative to the reference and there is no true SNP at this site. However, this individual was called as a het (C/T) because of the reads that end in the middle of the insertion. For these reads the penalty for a single mismatch to the reference is less than introducing a gap.

SNP calling II – Creating a reference for GATK and Picard
Using GATK and Picard was difficult because we had to keep modifying our reference (and therefore redo the alignments each time) to use these programs efficiently. Below I list the things we had to do to the reference to get them to work:
SNP calling I – alignment programs and PCR duplicates
I have tested various SNP calling methods using exome re-sequencing data from 12 interior spruce samples. I tried Bowtie2, BWA (mem), Picard (mark duplicates) and GATK for indel realignment and base quality recalibration. For SNP calling I used mpileup with and without BAQ as well as the Unified Genotyper from GATK. For an interesting and informative workshop outlining the Broad best practices SNP calling pipeline check out these youtube videos (http://www.youtube.com/watch?v=1m0ZiEvzDKI&list=PLlMMtlgw6qNgNKNv5V9qmjAxbkHAZS1Mf). My results are in a series of blog posts and I hope you find them useful. Please let me know if you have any suggestions for SNP calling. We only want to do the alignments and SNP calling once for the entire set of samples, because it is going to take a long time!
Estimating Insert Sizes
We recently had some trouble estimating insert sizes with our Mate Pair (aka Jumping, larger insert sizes) Libraries. All the libraries sequenced by Biodiversity and the Genome Sciences Centre (GSC) were shockingly bad, but the libraries sequenced by INRA were very good. For example, according to the pipeline, the GSC 10kbp insert size library had an average 236bp insert size, but the INRA 20kb library an average insert size of 20630bp.
See the histogram for the 10kbp library:
Filtering SNP tables
Previously I posted a pipeline for processing GBS data. At the end of the pipeline, there was a step where the loci were filtered for coverage and heterozygosity. How strict that filtering was could be changed, but it wasn’t obvious in the script and it was slow to rerun. I wrote a new script that takes a raw SNP table (with all sites, including ones with only one individual called) and calculates the stats for each loci. It is then simple and fast to use awk to filter this table for your individual needs. The only filtering the base script does is not print out sites that have four different bases called at the site.
Heterozygosity, read counts and GBS: PART 2
Subtitle: This time… its correlated.
Previously I showed that with the default ML snp calling on GBS data, heterozygosity was higher with high and low amounts of data. I then took my data, fed it through a snp-recaller which looks for sites that were called as homozygous but had at least 5 reads that matched another possible base at that position (i.e. a base that had been called there in another sample). I pulled all that data together, and put it into a single table with all samples where I filtered by:
C/C++ Dependency Troubleshooting
Unix C/C++ programs are very finicky about the compiler and library versions. Compiling is the process of translating human readable code into a binary executable or library that contains machine-friendly instructions. Linking is the process of telling multiple executables or libraries how to talk to each other.
gcc is a GNU C compiler that is typically used on unix systems. g++ is a GNU C++ compiler on unix systems. libstdc++ is the GNU standard C++ library. glibc is the GNU standard C library. When you install GCC on your unix machine, you are installing a package of the aforementioned items. The gcc unix command is smart enough to call either the gcc compiler or g++ compiler depending if you pass it C or C++ code.
If you attempt to run your program with an older standard library than it was originally linked it with, your program will crash and complain. Here are some tips to get around it.
C / C++ Troubleshooting
IN PROGRESS
Most of the high-performance bioinformatic programs are written in C or C++. Unfortunately, C and C++ code is some of the hardest code to debug. If you have only programmed casually in perl/python, you will not have a good time. Here are some tips to help you out, but you will most likely need someone with C / C++ programming experience and knowledge of the code to get you through it.
SmartmonTools & GSmartControl
Smartmontools is a command-line Hard Drive Diagnostic Tool that gives you clues on how long your disk has to live. You can run it manually, or you can configure it to periodically test your drives in the background and notify you about test failures via email.
GSmartControl is a GUI for Smartmontools and much easier to use.
Check out this Ubuntu SmartmonTools Tutorial on how to install and set them up.
Here are some tips that are not easily gleaned from the previous websites:
Continue reading
AWK unlinked SNP subsampling
For some analyses, like STRUCTURE, you often want unlinked SNPs. For my GBS data I ended up with from 1 to 10 loci on each contig which I wanted to subsample down to just one random loci per contig. It took me a while to figure out how to do this, so here is the script for everyone to use:
cat YOUR_SNP_TABLE | perl -MList::Util=shuffle -e ‘print shuffle(<STDIN>);’ | awk ‘! ( $1 in a ) {a[$1] ; print}’ | sort > SUBSAMPLED_SNP_TABLE
It takes you snp table, shuffles the rows using perl, filters out one unique row per contig using awk, then sorts it back into order. For my data, the first column is CHROM for the first row and then scaffold###### for the subsequent rows so the sort will place the CHROM row back on top. It might not for yours if you have different labels.
Text Editor Indenting & Highlighting
Do you hate reading code that is poorly indented or lacks syntax highlighting? Here are some common text editor fixes:
Continue reading
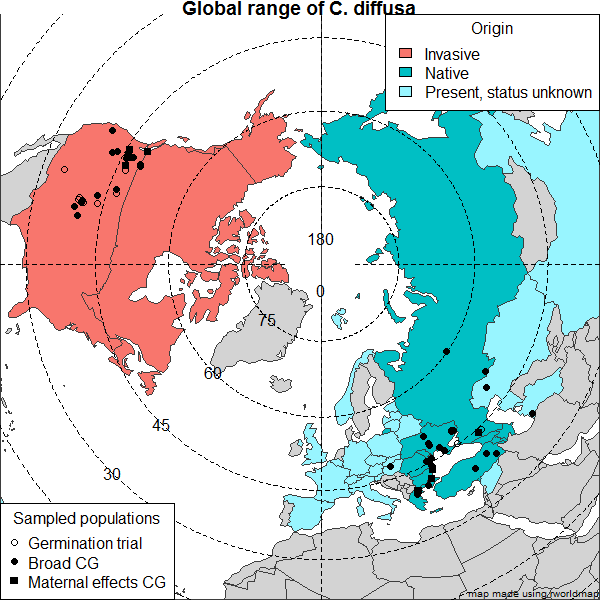
Modern map of Eurasia in R
How to Upload Files to Bitbucket (commandline)
BitBucket is an external source code repository server that hosts our shared code. Our repositories use Git as the version control program to keep track of file changes. The idea is you make changes to code on your local machine then share your code with everyone else by uploading to BitBucket.
The instructions below guide you step-by-step in uploading files to BitBucket using the commandline. Git is one of the most popular version control programs but it is not easy for beginners. If you want to do something that deviates from these steps, consult a git reference. Once you understand the basics of the git workflow, you can use a GUI program which can combine multiple steps in a single click.
Methods Write-up for Identifying Contamination in Sequencing Reads
Chris and I devised and implemented this method to identify contaminant DNA in our HA412 454 and Illumina WGS reads.
N.B. I will update this posts with links to the scripts once I get them up on the lab’s bitbucket account, plus extra details if I think they are necessary on review.
Blast2GO
This describes how you can run blast2go on a server using b2gpipe and a local database. This makes blast2go a viable option for annotating large fasta files. Otherwise it is much too slow. The database is currently set up on an AdapTree server. This took a while for me to troubleshoot, so you could run into different problems, but you will hopefully avoid some of the issues I ran into. The b2g Google group is good for troubleshooting. You can find many of these instructions at http://www.blast2go.com/b2glaunch/resources/35-localb2gdb
Allowed File Types At RLR – you can now upload scripts with their usual file extensions
Hello All,
Many of us have been annoyed by the restricted file types that WordPress allows to be uploaded to RLR. It’s especially annoying because all WordPress is doing when it permits or denies an upload is checking the file extension against a list of allowable extensions. {Even the most malicious code could be uploaded to our blog as long as it had a .txt file extension. Whether that code could then be made to execute, however, is far beyond my web-programming grasp – WordPress would treat it as plain text so it may be impossible.}
We’ve been sharing code via RLR by sidestepping the file extension rules and uploading scripts as .txt text files or by compressing files into zip archives or just putting the code itself into posts. Admittedly these were simple solutions, but now it’s even simpler – I just added some of the relevant file extensions to the list that RLR will allow for upload.
I added: “.pl”, “.py”, “.sh”, “.R”, “.r” and “.kml”.
Any file with one of those extensions will upload as plain text, i.e. WordPress will treat it as a text file.
If I’ve omitted something useful let me know.
Please remember that code can simply be copied into the body of a post and that will often be the best way to share it. But, in addition to that presentation, and especially for long scripts, you can now upload the script with its file extension to the RLR media library and put a link to it in your helpful post explaining what it does.
Dan.
Text File To kml – Perl Script
Google Earth reads and writes a special form of xml file called a kml (keyhole markup language). Many other geographic viewers and GISs can also read kml files so it’s not a bad thing to be able to make kml files for sample location data. I assume there are many ways to do this. The way I have done it is via a perlscript that I wrote. This post provides that script and explains what it does.
Here is the script, its called texttokml.pl.
It’s very simple and I commented it heavily so even the most naive perl programmer should be able to figure it out and change it but if you want me to hold your hand just ask.
Explanation follows . . .
Scripts for Formatting SNP Tables
Some SNP table to useful table conversion scripts are here: FormattingScripts_v0.4
Readme.txt explains usage, makes fasta, bayescan, structure files as well as converting to digits for R.
Let me know if you find any of this useful or broken.
Greg
Edit: updated small fix to structure formatter