
As you know, the sunflower genome contains a large amount of repetitive sequences, that is why it is so big and so annoying to sequence. I have been working for a while on optimizing a depletion protocol, to try to get rid of some repetitive sequences in NGS libraries (transposons, chloroplast DNA…). Continue reading
Category Archives: Lab Bench
Quick update: 96-well plate CTAB DNA extraction with fresh tissue
When I posted the protocol I have been using for CTAB DNA extraction in 96-well plates, I included results from a few plates I did starting from dried H. anomalus leaves I collected a couple of months earlier in Utah. While they showed that the method worked well enough when starting from “difficult” material, they were not exactly what you’d dream of when you decide to extract DNA, especially if you are starting instead from fresh material.
Here are the results from a plate of extraction I did starting from individual small (1.5-2 cm in length), young leaves from ~3 month-old H. anomalus plants. I collected the leaves directly in 96-well plates (I already put one metal bead in each well), put them on dry ice until I got to the lab (a couple of hours), left them overnight in the -80, and started extracting DNA the day after.
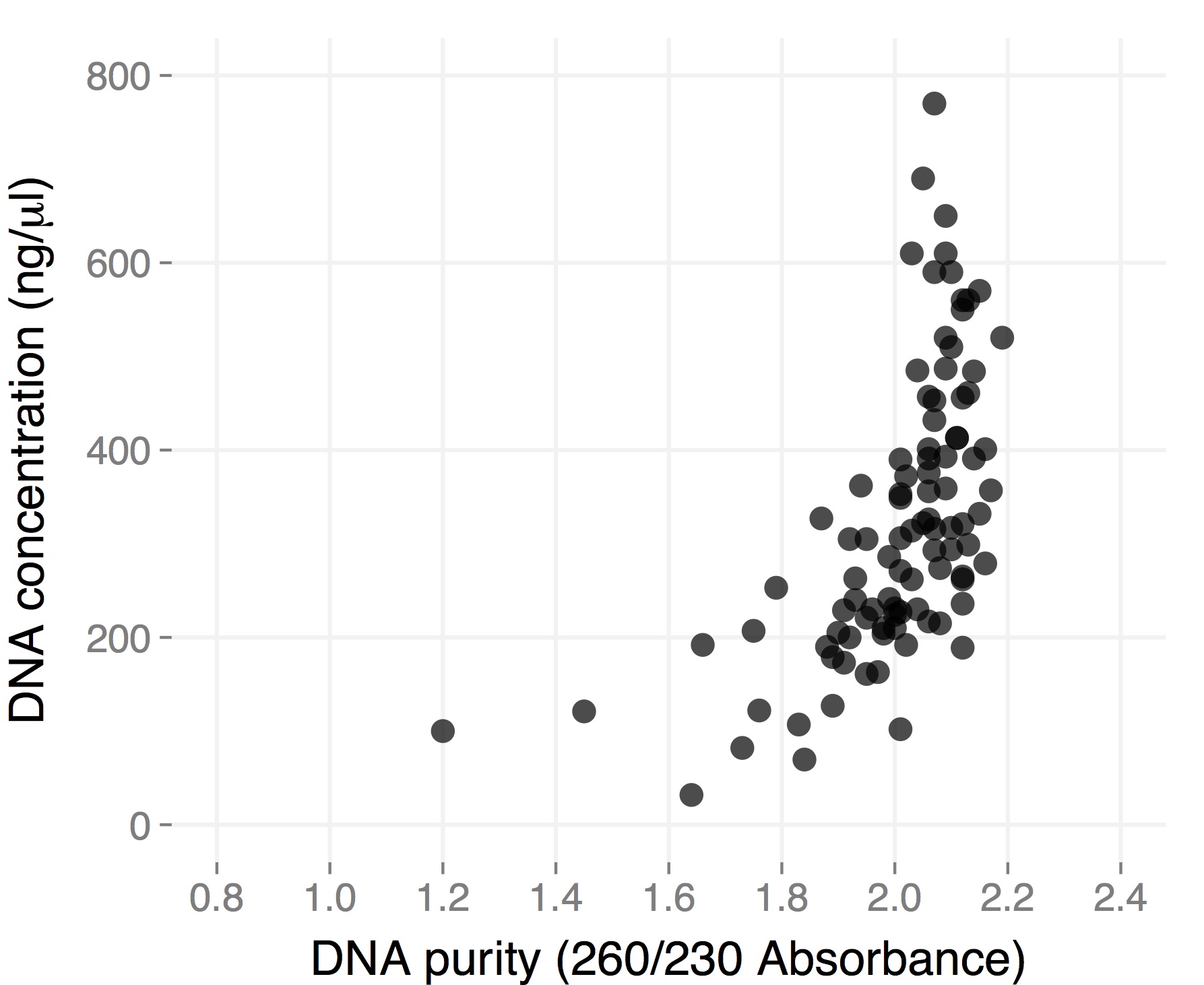
The final volume was 50 microliters, so total yield is for most samples between 10 and 30 micrograms of DNA. These are “real” DNA concentration measured by Qubit. Both average yield and purity are considerably higher than for dry tissue, and they are comparable to what you would get starting with frozen tissue using the single tube protocol (but you save a ton of time). Hope this gets you all more thrilled about 96-well plate DNA extractions 🙂
Where does all the GBS data go? Pt. 2
An analysis aimed at addressing some questions generated following discussion of a previous post on GBS…
Number of fragments produced from a ‘digital digestion’ of the Nov22k22 Sunflower assembly:
Clai: 337,793
EcoRI: 449,770
SacI: 242,163
EcoT22I: 528,709
PstI: 129,993
SalI: 1,210,000
HpaII/MspI: 2,755,916
Here is the size distribution of those fragments (omitting fragments of >5000bp):
All the enzymes
With Msp removed for clarity
Take home message: PstI produces fewer fragments of an appropriate size than other enzymes. It looks like the correlation between total number of fragments and number that are the correct size is probably pretty high.
Now for a double digestion. Pst and Msp fragment sizes and again omitting fragments >5000bp. This looks good. In total fragment numbers (also omitting >5000bp fragments):
Pst+Msp total fragments: 187271
Pst+Msp 500<>300bp: 28930
Pst alone total: 79049
Pst alone 500<>300bp:6815
Take home: Two enzyme digestion could work really well. It may yield more than 4 times more usable fragments. I do think we could aim to get even more sites. Maybe some other RE combination could get us to the 100,000 range. With a couple of million reads per sample this could still yield (in an ideal world) 10x at each site. Send me more enzymes sequences and I can do more of the same.
Edited for clarity and changed the post name
Where does all the GBS data go?
Why do we get seemingly few SNPs with GBS data?
Methods: Used bash to count the occurrences of tags. Check your data and let me know what you find:
cat Demutliplexed.fastq | sed -n '2~4p' | cut -c4-70 | sort | uniq -c | grep -v N | sed 's/\s\s*/ /g' > TagCounts
Terminology: I have tried to use tags to refer to a particular sequence and reads as occurrences of those sequences. So a tag may be found 20 times in a sample (20 reads of that sequence).
Findings: Tag repeat distribution
Probably the key thing and a big issue with GBS is the number of times each tag gets sequence. Most sites get sequenced once but most tags get sequence many many times. It is hard to see but here are histograms of the number of times each tags occur for a random sample (my pet11): All tags histogram (note how long the x axis is), 50 tags or less Histogram. Most tags occur once – that is the spike at 1. The tail is long and important. Although only one tag occurs 1088 times it ‘takes up’ 1088 reads.
How does this add up?
In this sample there are 3,453,575 reads. These reads correspond to 376,036 different tag sequences. This would mean (ideally) ~10x depth of each of the tags. This is not the case. There are a mere 718 tags which occur 1000 or more times but they account for 1394905 reads. That is 40% of the reads going to just over 700 (potential) sites. I’ve not looked summarized more samples using the same method but I am sure it would to yield the same result.
Here is an example: Random Deep Tag. Looking at this you can see that the problem is worse than just re-sequencing the same tag many times but you introduce a large number of tags that are sequencing and/or PCR errors that occur once or twice (I cut off many more occurances here).
Conclusion: Poor/incomplete digestion -> few sites get adapters -> they get amplified like crazy and then sequenced.
Update 1:
Of the > 3.4Million tags that are in the set of 18 samples I am playing with only 8123 are in 10 or more of the samples.
For those sites with >10 scored the number of times a tag is sequenced is correlated between samples. The same tags are being sequenced repeatedly in the different samples.
Update 2:
As requested the ‘connectivity’ between tags. This is the number of 1bp miss matches each tag has. To be included in this analysis a tag must occur at least 5 times in 3 individuals. Here is the figure. So most tags have zero matches a smaller number have one and so on. This actually looks pretty good – at least how I am thinking about it now. This could mean that the filtering criteria works. If errors were still being included I would expect tags with one match (the actual sequence) to occur more than ones with zero.
96-well plates CTAB DNA extraction
When I was working with Arabidopsis, 96-well CTAB DNA extraction was my best friend, and I spent many days extracting away tens of thousands of samples. Good times.
DNA extraction is much less pleasant in sunflower, but since I was reasonably happy with the results of single-tube 3% CTAB DNA extractions, I though I would try to scale it up to a 96-well plate format. Results of earlier attempts, with the participation of Brook and Cris, ranged from inconsistent to disastrous. Things all but improved when I tried again after coming back from Utah with a few hundreds dried samples. Since though the prospect of extracting them all one by one didn’t sound very attractive, I put some more effort into improving the protocol, and now it works quite nicely.
Plant DNAzol
Hi all,
Here’s another method for DNA extraction. The blog is stuffed to the gills with DNA extraction methods. The current standards are ‘Qiagen-like’ columnless – used to generate the DNA for the genome sequencing project and CTAB. I add this protocol because itis
easy and it is effective.
I extracted from Ha89 and harvested ~ 115 ng/uL from 20 cm tall plants. I also purposely ‘took it slow,’ letting tissue thaw after freezing to see PlantDNAzol’s efficacy. It’s efficient.
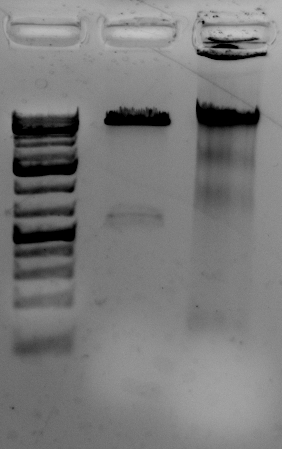
Quality is good
The gel to the left:
Right most lane contains Ha89 genomic DNA – 10 uL loaded of 70 at 115 ng/uL DNA – 260/280 was 1.78. 260/230 was ~1.00 (I suspect I could have added an additional ethanol wash to remove Guandine from the DNA mixture.
Is the DNA useful? Can downstream reactions proceed?
Yes. I digested the DNA with a methylation sensitive restriction enzyme, PstI and a methylation insensitive enzyme, EcoRV
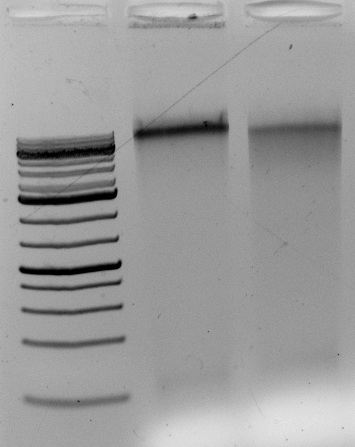
<–The gel to the left :
Leftmost lane Ladder
A vector digested with PstI,
Ha89 gDNA digested with PstI 240 minutes,
Ha89 gDNA digested with EcoRV 240 minutes.
The DNA digests. But does it contain contaminants that upset the enzymes over long incubations? Overnight?
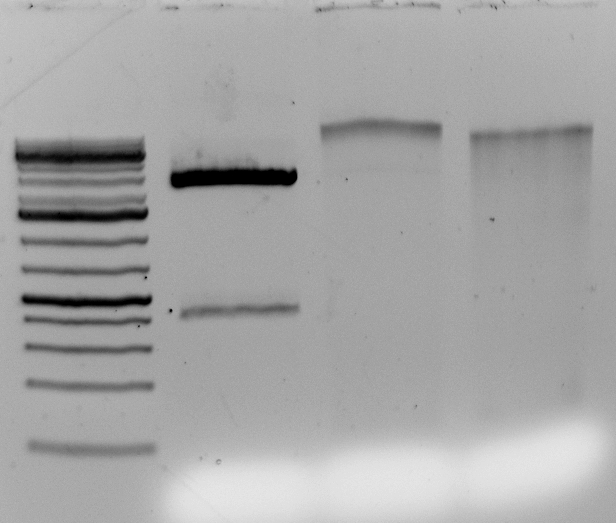
Gel above: From left- Ladder, unrelated vector digest,
Ha89 gDNA digested with PstI 22 hours
Ha89 gDNA digested with EcoRV 22 hours.
Protocol for DNAzol extraction (exactly as published by LifeTech but easier to follow – http://tools.lifetechnologies.com/content/sfs/manuals/10978.pdf:
Have these items on hand:
1 0.6 mL DNAzol per 100 mg sample
2. 0.3 mL chloroform per 100 mg sample
3. Timer
4. 100% ethanol (0.225 mL per sample),
5. 75% ethanol (0.3 mL per sample)
Handle all inversions carefully. When you see invert or shake handle your samples gently
1. Mix 100 mg ground tissue with 0.3 mL PlantDNAzol – 100 mg is max. Overdoing will hurt your yield.
2. Invert gently to aid in lysis and dispersion
3. Once completely dispersed incubate at RT, 5 min, shake periodically.
4. Add 0.3 mL chloroform and mix.
5. Once completely dispersed incubate at RT, 5 min, shake periodically.
6. Centrifuge at RT, 12 000 g (NOT rpm) 10 min
7. Harvest the supernatant. – you’ll see a phenol/chloroform styled triple layer. The middle layer will be pulpy containing your cellulosic debris and proteins. Don’t collect the middle layer. Less is more
8. Mix supernatant from 7 with 225 uL 100% ETOH.
9. Incubate at RT, 5 min
10. Centrifuge mixture 5000 g 4 min – get preparing for step 11
11. Make a PlantDNAzol – Ethanol mixture: For one sample mix 0.3 mL PlantDNAzol with 0.225 mL 100% ethanol.
12. Discard the supernatant from 10 and mix it with 0.3 mL of the mixture prepared in step number 11
13. Incubate as in step 9.
14. Spin as in step 10.
15. Pour off supernatant
16 Wash pellet with 75% ethanol – 0.3 mL – this step can be repeated if your 260/230 isn’t adequate. Guanidine absorbs strongly in 230 nm wavelength
EDIT: repeat step 16 for a total of 2 washes.
17. Spin as in step 10.
18. Remove supernatant – if your samples are green repeat step 16.
19. Resolubilize your DNA in TE or NaOH. Make sure to run your bead of TE over the wall of the tube to collect your DNA.
EDIT – Less is more as is usually the case with DNA extraction. I harvested from 38, 60, and 100 mg of tissue. – A sweet spot for tissue quantity is 45 to 60 mg for the given amount of Plant DNAzol
Qubit quantifiication: 45 mg of tissue yielded 264 ng/uL ug/mL. 60 mg, 305 ng/uL. 100 mg, 12.4 ng/uL
Normalize/quantify your WGS libraries
Regardless of what method you use to make your Illumina libraries, if you added barcodes or indices you will need to normalize them before pooling (or otherwise have probably very uneven coverage). Or, you might want to know the exact molarity of your library before sending it for sequencing (although the need for that in our case is debatable, see later). The most accurate way to do both is probably by qPCR. Continue reading
(Probably the closest you can get to) Home-brew Illumina WGS libraries
As some of you might know, I have been working for the last few months on optimizing a protocol for Illumina WGS libraries that will reduce our dependency on expensive kits without sacrificing quality. The ultimate goal would be to be able to use WGS libraries as a more expensive but hopefully more informative alternative genotyping tool to GBS. Getting to that point ideally requires to develop:
1) A cheaper alternative for library preparation (this post)
2) A reliable multiplexing system (this other post)
3) A way to shrink the sunflower genome before sequencing it (because, as you know, it’s rather huge) (yet another post)
The following protocol is for non-multiplexed libraries. The protocol for multiplexed ones is actually identical, you just need to change adapters and PCR primers – more about that in the multiplexing post.
If you are planning to pool libraries and deplete them of repetitive elements, read carefully all three posts before starting your libraries (mostly because you might need to use different adapters and PCR primers)
Tissue dessication with table salt
There is a new paper published by Elena Carrió and Josep A. Roselló online early in Molecular Ecology Resources that suggests salt dessication of leaves dehydrates and prevents decay at levels similar to that of silica gel, with similar PCR results.
Large-grain silica is probably still the best option, but this would come in really handy if you come across something interesting that you want to collect but don’t happen to have silica gel with you.
Here is the main figure (link to the paper below):

Thanks to Maggie Wagner from the TMO lab who found this paper and sent it around. Here is a link to the full article:
http://onlinelibrary.wiley.com/doi/10.1111/1755-0998.12170/full
DOI: 10.1111/1755-0998.12170
GBS items – discount prices negotiated with Invitrogen
Hi everyone,
I met with Invitrogen and was able to negotiate a price break for PstI and T4 ligase
Are there other suggestions for items we can add to this quote?
Are people still doing their library Q.C. with realtime PCR?
Invitrogen PstI and T4 DNA ligase

PstI digest tests
Edit: I forgot to thank DanB and Kate for the generous donation of DNA. Sorry guys 🙁
Edit 2: After the lab meeting some questions have been asked.
To summarize :
1. All the enzymes work equally well. There is a slight performance decrease if one uses the PstI-HF.
2. I’d recommend using the PstI from Invitrogen – Invitrogen works extremely well and is the cheapest among the Psts tested ( $22 versus $75 ). EDIT: Brook and Kate let me know that I failed to factor in the units provided for a given dollar value. Invitrogen provides the cheapest enzyme.
For NEB – With our volume purchases we get 10000U for $71.40.
For Invitrogen – With our volume purchases we get 10000U for $63.90 {represents price negotiated with Helen, accounts manager at Invitrogen Stores}
Thanks guys for pointing that out!
This adds up considering how much we’re going through.
3. Sunflower gDNA does not digest completely in 3 hours. It is recommended to go overnight or 18 hours with your digestion.
4. After 18 hours do not be alarmed by incomplete digestion. This is OK according to RFLP work performed by Loren. There should be a sufficient number of fragments for GBS libraries.
Hi all,
Here are the results from the PstI digest tests:
All enzymes tested fail to fully digest sunflower gDNA.
Enzymes from Invitrogen, Thermofisher/fermentas, NEB (non HF) and NEB HF were tested. All performed equivalently. I would say the NEB HF was slightly less processive after 70 min. See attached PDF for gels and full documentation of reaction conditions.
**** 2013-Oct-08-debono-digesttest *****
Thanks
Allan
NEB HF enzymes – regarding library preps
Hi guys,
Just a note about PstI-HF:
The PstI-HF sold from NEB is less processive than the old NEB PstI/red stripe version (go here to see it: https://www.neb.com/products/r0140-psti) . When digesting ultrapure, high concentration homogeneous DNA (plasmids) digestions fail to go to completion even when left overnight (> 16 hours). What does this mean for you? It means that your genomes which are more difficult substrates to deal with will not digest fully unless left to go overnight. If you want to ensure a good digest use a positive control and switch to the linked product.
How my PhD was saved by DTT, or: how to get moderate quantities of clean, unsheared DNA from tricky tissues
I have been trying for months to find a high-throughput solution to DNA extraction from lyophilized (freeze-dried) leaf discs of Helianthus argophyllus, a species known for its intransigent polyphenolics and low DNA yields. And I appear to have found one, thanks to Horne et al. 2004 (here) and Dow Chemical!
Lab Safety training
UBC offers two levels of safety courses.
For those who work under supervision (i.e. undergrads), there is a basic online course found here http://riskmanagement.ubc.ca/courses/intro-lab-safety
This is a requirement for all undergrads who will be working in the lab. Once your student has taken the course, it is expected that you (their supervisor) will follow up with reviewing lab safety training specific to our lab. Please remember that these students need supervision at all times when working in the lab.
For lab personnel who do not require supervision there is a more advanced course, http://riskmanagement.ubc.ca/courses/laboratory-chemical-safety, it includes an online and practical component. This course is mandatory, unless you’ve had similar training at a previous institution. Megan will provide in lab follow-up for new personnel to cover safety features of the lab.
UBC also offers other specific training, although the courses aren’t likely relevant to the type of lab work we do; such as Radionucleotides, and biohazards, please see http://riskmanagement.ubc.ca/courses.
GBS, coverage and heterozygosity
I’m running some tests on my GBS data to look for population expansion. I know from looking at GBS data from an F1 genetic mapping population that for GBS data heterozygotes can be under called due to variation in amplification and digestions. Also, for my data observed heterozygosity is almost always under expected. Heterozygotes can also be overcalled when duplicated loci are aligned together. The tests I’m going to use explicitly use observed heterozygosity so this is worrying.
Ph*cking phenolics!
Hi guys,
I have extracted sunflower DNA a dozen times now. I get one consistent problem a gelatinous film coprecipitating with my DNA (carbohydrates/polysaccharaides). I also get sharp smelling phenolic compounds. These occur regardless of DTT/2-BME addition.
I found a paper detailing the use of 2-butoxyethanol (2-BE) to remove strawberry polysaccharides and phenolics. The researcher’s idea is to perform a two-step precipitation:
Silica gel: God’s gift to botanists
2
Another Rieseberg lab member was asking me this week about preserving plant tissues on silica gel for later DNA extraction, and this got me to thinking about the general idea. I thought I would make a post about it, since it’s a useful technique, even in the genomic age. A lot of you already know all about this, so apologies for preaching to the choir.

The miracle of silica gel
I put an extensive post on my own Research Blog, but here is the gist of it:
Chemical safety: a quick read
Chemical safety: a quick read
Hi everyone,
Here’s a sobering blog post I saw at BiteSizeBio. I use most of these chemicals regularly. Scarier still I had no idea chloroform becomes phosgene once metabolized. The list contains popular chemicals in the Rieseberg lab.
original link: http://bitesizebio.com/articles/ten-bad-chemicals-in-the-lab-and-what-they-do-to-you/
Ten Bad Chemicals In The Lab and What They Do To You!
SPRI beads (almost) for free!
A few weeks ago Kristin posted a nice blog entry on home made SPRI beads, which effectively replace the ridiculously expensive commercial AMPure beads. (Editor’s note: see this post at RLR for more about the AMPure beads). Dan already ordered the ingredients to make them in our lab as well, and when they all arrived I volunteered to prepare and test them. Following are some results. If you don’t feel like reading through the whole post, the bottom line is that they work very well, and that you are very welcome to use them (they are in an aluminium-wrapped Falcon tube in the fridge, with a white “SPRI beads” label).
Normalizing DNA with the Robot
Im not looking to volunteer myself as the robot expert, this is just one of the things you need if you are going to use it. These are the files needed to normalize a plate of DNA. One instructs it to take the volumes of DNA from one plate and put them into another and the other is to add the water. I can also offer a poor picture of the machine itself.
