
Genetic architecture and genomics of ecological speciation with gene flow
Threespine Stickleback (Gasterosteus aculeatus complex) species pairs. Photo by Matt Arnegard.
While thought for many years to be practically impossible, ecological speciation with gene flow now appears to be common. How co-adapted differences evolve and remain associated with one another despite interbreeding with the opposite type though, has been an intellectual challenge for decades. Dolph Schluter and I have written a review article of the genetics of ecological speciaton (Schluter and Conte. 2009. PNAS 106: 9955-9962).
In sticklebacks:
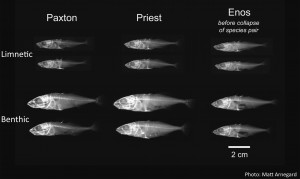
The recent and repeated ecological speciation of threespine stickleback species pairs that co-occur in lakes, has become a model system for investigating such questions. For my PhD research with Dolph Schluter, I use these repeatedly evolved benthic and limnetic species pairs to investigate the genetic architecture of reproductive isolation between them. In doing so, I aim to better understand the genetic parameters under which ecological speciation with gene flow may occur and how likely the genetics of ecological speciation with gene flow are to be repeated in independently evolved species.

Our study involves collecting phenotype data from multiple F2 populations raised in the UBC experimental ponds for QTL mapping. Pictured here is assistant, Jacob Best, collecting a breeding male at his nest. Photo by Gina Conte.
I am working with a team of collaborators including, Dolph Schluter, Katie Peichel and Matt Arnegard to answer these questions. We conducted multiple large-scale QTL mapping experiments in simulated natural conditions to map the genetic basis of extrinsic post-zygotic reproductive isolation and assortative mating in multiple benthic and limnetic species pairs. The first study of these QTL mapping studies was recently published (Arnegard et al. 2014. Nature 511: 307-311). We mapped the genetic architecture of juvenile niche divergence in the Paxton Lake species pair. This niche divergence axis explained a valley shaped growth rate landscape, whereby the most limnetic–like and benthic-like F2 hybrids had the highest growth rates, and the most intermediate F2 hybrids had the lowest growth rates. We found that niche divergence was explained by multiple, unlinked QTL. Interestingly, this genetic architecture appeared to be largely additive, with each benthic allele making an approximately equal contribution to an overall benthic niche phenotype. Studies to estimate the genetic architecture of prezygotic isolation between the benthic and limnetic species pairs are still ongoing. In these studies, we are attempting to map the loci that contribute to differences in female mate preference and in male nesting habitat choice. Articles describing the results of these studies are soon forthcoming.
In addition, I have carried out a manipulative experiment that showed that both benthic and limnetic stickleback females from Paxton Lake prefer mates whose body size more closely matches their own (Conte and Schluter. 2013. Evolution 67(5): 1477-1484). This result has implications for the genetics of divergence between the species pair. Along with previous research, it suggests that the genes that control body size are 1. under divergent selection, 2. control mate signal and 3. determine mate preference. Thus, divergent selection in body size should lead to the evolution of assortative mating by body size as an automatic by-product, unhindered by recombination. The genetics of divergence between the Paxton lake species pair, in this one way, are extremely favorable for the speciation process.

Pea Aphid (Acyrthosiphon pisum)
photograph © Alex Wild 2003
In pea aphids:
With Sara Via, I studied two incipient species of pea aphids in the Eastern U.S. that are specialized for use of different host plants (alfalfa and clover), and divergence persists despite gene flow between them. With a QTL map of fitness proxies (performance and fecundity) on the alternate host plants in hand, we performed a genome scan for Fst outliers. The combination of a QTL map and a genome scan allowed us to test key hypotheses about the effects that divergently selected loci have on loci surrounding them (Via et al. 2012. Mol. Ecol. 21(22): 5546-5560). We found evidence for large genomic regions of reduced gene exchange around QTL for performance and fecundity, consistent with the hypothesis that ‘divergence hitchhiking’ is occurring.
The predictability of the genetic basis of adaptation
One of the most important and instructive questions currently being asked within the field of evolutionary genetics is: ‘how predictable are the genetics of adaptation?’ Studying the predictability of the genetics of adaptation leads to understanding the core forces shaping them. A promising way to estimate how predictable the genetics of adaptations are is to look at how commonly repeated phenotypic adaptation is underlain by repeated genetic evolution. We conducted a meta-analysis to estimate the probability of parallel and convergent genetic evolution across broad diversity of natural populations (Conte et al. 2012. Proc. R. Soc. B 279: 5039-5047). Using an impartial literature review, we estimated the probability of gene ‘reuse’ in natural populations undergoing repeated phenotypic evolution to be 0.32-0.55, depending on the genetic methods employed by individual studies. Furthermore, we found that the probability of gene reuse declines with increasing age of the taxa being compared.
To obtain more a comprehensive estimate within a system, we measured the extent of genetic parallelism underlying parallel morphological divergence between the Paxton and Priest Lake species pairs of threespine stikcleback (Conte et al. 2014. submitted). We found that about 50% of QTL for parallel morphological differences are parallel, and on average, the proportional similarity of QTL use underlying individual morphological traits is about 0.4. In addition, we find no evidence that the phenotypic effect size of QTL predicts whether or not they are parallel. These represent the most comprehensive estimates to date in a single system, providing further insight to the predictability of the genetics of adaptation.

Large numbers of plants are being genotyped and phenotyped in six environments. Photo from AdapTree website.
AdapTree
AdapTree is a large interdisciplinary collaboration intended to “assess the adaptive portfolio of reforestation stocks for future climates”. We are doing this though large-scale studies of population and landscape genomics in lodgepole pine (Pinus contorta) and interior spruce (Picea glauca and Picea engelmannii hybrid complex). These tree species are two of the most commonly logged in western Canada. A primary goal of the project is to discover the genetic basis of climate adaptation using phenotype and environmental data along with an enormous amount of genomic data from an exceptional number of populations and individuals collected across the species ranges. These results will not only make a significant contribution to the field of evolutionary genomics, but will also be directly used by tree breeders to ensure that reforestation stocks will be well adapted to current and future climates. Although I am involved in many aspects of the AdapTree project, one study I am particularly focused on is attempting to quantify the amount of genetic load that exists in local populations, how it varies across the range and how it relates to local climate adaptation.
Evolution of sexual signalling in an endemic Hawaiian cricket (Laupala) radiation

Laupala makaio and L. paranigra paired in a mating trial. Photo by Gina Conte.
In Kerry Shaw‘s lab, I studied a large and rapid radiation of endemic Hawaiian swordtail crickets of the genus Laupala, that has occurred by successive colonization from older to younger Hawaiian islands, followed by diversification within islands. These species are primarily differentiated by secondary sexual signals including the pulse rate of the male courtship song, and the size of male genitalia. Migration between islands is thought to be sufficiently rare that colonization of new islands were likely to constitute founder events. Thus, we tested ‘Kaneshiro’s hypothesis’, which predicts that elements of male courtship could be lost in founding populations due to genetic drift coupled with relaxed sexual selection (Oh et al. 2013. Curr. Zool. 59 (2): 230–238). We found asymmetrical sexual isolation between an ancestral species from Maui, Laupala makaio, and a derived species from Hawaii, Laupala paranigra, in the direction predicted by Kaneshiro’s hypothesis. This result strengthens that of a previous study that found asymmetrical sexual isolation in another interisland, ancestral-derived pair, but for which male pulse rate was a confounding variable.