
I’m running some tests on my GBS data to look for population expansion. I know from looking at GBS data from an F1 genetic mapping population that for GBS data heterozygotes can be under called due to variation in amplification and digestions. Also, for my data observed heterozygosity is almost always under expected. Heterozygotes can also be overcalled when duplicated loci are aligned together. The tests I’m going to use explicitly use observed heterozygosity so this is worrying.
As a first pass at this, I’ve compared observed heterozygosity to coverage. For this, I took GBS data from two lanes of H. exilis (192 samples). As a proxy for read number, I used file size of the demultiplexed fastq file of both reads together. They were demultiplexed using the Baute/Rose GBS demultiplexer. This does not support mismatches in the barcode sequence. Sequences were aligned to the Nov22k22 Annuus reference using Bowtie2. SNPs were called using the lab’s maximum likelihood script. I didn’t filter those scripts, I just measured observed heterozygosity (#heterozygotes/#calls that are not NN).
I plotted this and then fit a loess curve through the data. Black dots are the first library, red are the second. The second library was broader (300-600 bp vs 350-550 bp).
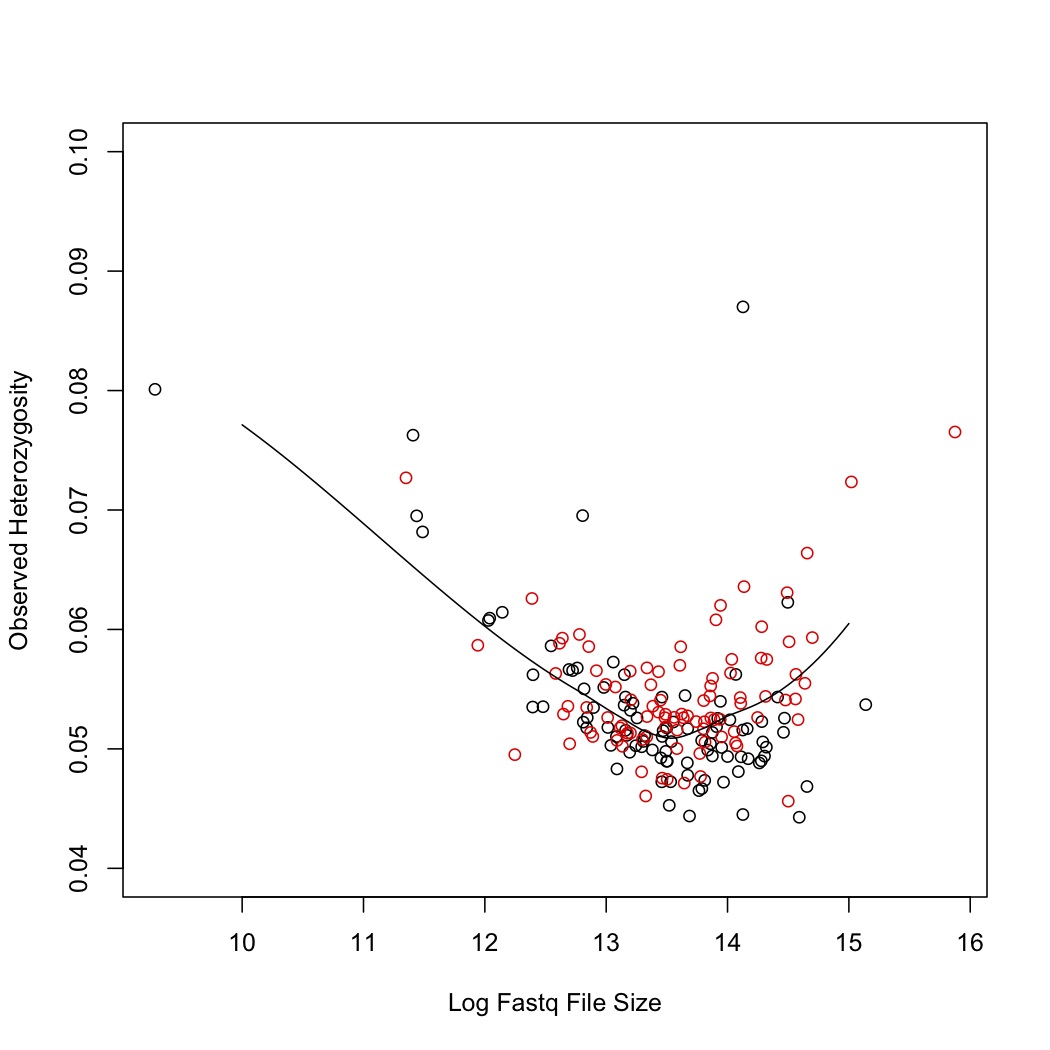
I was expecting a result where more reads increased heterozygosity because it would reduce false homozygotes. That might be the case at the really high end, but there is also a clear pattern of increased heterozygosity at the low end of read number.
This could be because GBS read depth is often skewed. So for a true heterozygote you might get a read depth of 20 A, 400 T. With the current ML SNP caller, these are treated as homozygotes. Perhaps higher read counts just exaggerate this problem.
Although this is worrying, it does seem that within the normal range (in my dataset it corresponds to Fastq files from ~200M to 1.5G), it is fairly even.
This is something those of us using GBS data should keep in mind.